ABSTRACT
E. coli YdbK is predicted to be a pyruvate:flavodoxin oxidoreductase (PFOR). However, enzymatic activity and the regulation of gene expression of it are not well understood. In this study, we found that E. coli cells overexpressing the ydbK gene had enhanced PFOR activity, indicating the product of ydbK to be a PFOR. The PFOR was labile to oxygen. The expression of ydbK was induced by superoxide generators such as methyl viologen (MV) in a SoxS-dependent manner after a lag period. We identified a critical element upstream of ydbK gene required for the induction by MV and proved direct binding of SoxS to the element. E. coli ydbK mutant was highly sensitive to MV, which was enhanced by additional inactivation of fpr gene encoding ferredoxin (flavodoxin):NADP(H) reductase (FPR). Aconitase activity, a superoxide sensor, was more extensively decreased by MV in the E. coli ydbK mutant than in wild-type strain. The induction level of soxS gene was higher in E. coli ydbK fpr double mutant than in wild-type strain. These results indicate that YdbK helps to protect cells from oxidative stress. It is possible that YdbK maintains the cellular redox state together with FPR and is involved in the reduction of oxidized proteins including SoxR in the late stages of the oxidative stress response in E. coli.
INTRODUCTION
Reactive oxygen species (ROS) are generated in living cells as inescapable by-products of incomplete reduction of molecular oxygen during normal cellular metabolism (Cui et al., 2012). The generation of ROS is enhanced by the exposure of cells to exogenous stimuli such as ionizing radiation and various chemical oxidants (Cui et al., 2012). Cellular components such as DNA, proteins and lipids are easily oxidized and inactivated through oxidation by ROS (Cui et al., 2012). DNA damage generated by ROS has been implicated in mutagenesis, carcinogenesis and aging (Cui et al., 2012; Klaunig et al., 2011). To overcome these deleterious effects, bacteria and eukaryotic cells have evolved defense mechanisms against oxidative stress caused by ROS (Imlay, 2008; Lushchak, 2011).
When exposed to excess ROS, cells respond by expressing a specific set of genes. A bacterial superoxide stress sensor, SoxR, is constitutively expressed but inactive in its reduced form (Li and Demple, 1994; Pomposiello and Demple, 2001). When its [2Fe-2S] cluster is oxidized, SoxR transcriptionally activates the soxS gene (Wu and Weiss, 1991; Gaudu and Weiss, 1996). SoxS is a transcriptional activator of the AraC/XylS family and induces the expression of a large number of regulon genes that play critical roles in protecting cells from oxidative stress (Wu and Weiss, 1991; Pomposiello et al., 2001). SoxS-activated genes include superoxide scavenging enzyme (sodA), DNA repair enzyme (nfo), ferredoxin (flavodoxin)-NADP(H) reductase (fpr), transcriptional repressor for iron uptake (fur), oxidative stress-resistant isozymes of the TCA cycle (fumC, acnA) and efflux pump (acrAB) (Pomposiello et al., 2001; Martin and Rosner, 2002). As a negative-feedback mechanism, SoxS interferes its own expression by binding of excess SoxS proteins to the promoter region of the soxS gene (Nunoshiba et al., 1993). When superoxide stress diminishes, SoxR is reduced by NAD(P)H-dependent reductases (Kobayashi and Tagawa, 1999; Koo et al., 2003), and SoxS is degraded by proteases such as Lon (Griffith et al., 2004). The SoxRS regulon is known to be activated by the superoxide radical, but recent studies show that SoxR is also activated in the absence of superoxide when the cellular redox balance is disrupted (Krapp et al., 2002; Gu and Imlay, 2011). So, it is important to examine whether the SoxRS regulon includes certain reductases to restore and maintain the cellular redox state.
We previously isolated several novel superoxide-inducible (soi) genes in E. coli (Mito et al., 1993). Among these soi genes, soi-9 was identified to be the ydbK gene. A homology search predicted that YdbK is a putative pyruvate:flavodoxin oxidoreductase (PFOR) which catalyzes the oxidation of pyruvate to acetyl-CoA and CO2 and delivers electrons to flavodoxin (Serres et al., 2001). Flavodoxin is an electron transfer protein with FMN prosthetic groups and often substitutes for ferredoxin, another electron transfer protein with an iron-sulfur center, indicating that flavodoxin can also act as a substrate for the NADPH:ferredoxin oxidoreductases (Osborne et al., 1991). The ydbK gene is constitutively expressed in SoxS-overexpressing E. coli strains (Mito et al., 1993; Fabrega et al., 2012). E. coli ydbK mutants are highly sensitive to superoxide-generating agents (Mito et al., 1993; Eremina et al., 2010), indicating that YdbK plays a critical role in the defense against oxidative stress in E. coli. Furthermore, most PFORs are oxygen-labile (Williams et al., 1987; Wahl and Orme-Johnson, 1987; Kunow et al., 1995). Previously, it had been shown that ydbK gene and downstream ompN gene are co-expressed in the same mRNA transcript and activated by SoxS, and the authors suggested that this SoxS-dependency could be indirect via an unknown factor (Fabrega et al., 2012).
In this study, we found that ydbK was superoxide-inducible and obtained evidence for direct regulation of the gene expression by SoxS. We also found a critical role of YdbK in defense against oxidative stress. It is possible that YdbK and FPR function in restoring reducing equivalents in the late stages of the superoxide-induced response.
MATERIALS AND METHODS
Bacterial strains and plasmids
E. coli cells were grown in Luria-Bertani (LB) medium at 37℃ with vigorous shaking. When necessary, ampicillin (Ap), kanamycin (Km) and chloramphenicol was added to the medium at 100, 50 and 30 μg/ml, respectively. E. coli BW25113, its derivatives and plasmid clones pNT3-ydbK, pNTR-SD and ASKA soxS were supplied by National BioResource Project. A plasmid pMal-stop was constructed from pMal-c2 by introducing a stop codon in front of the lacZ coding region using oligonucleotides listed in Table 1. Other E. coli K-12 strains and plasmids used in this study are listed in Table 2 and Table 3, respectively (Carlioz and Touati, 1986; Nunoshiba and Demple, 1993; Datsenko and Wanner, 2000; Baba et al., 2006; Casadaban et al., 1980).
Table 1. Oligonucleotides used in this study
| Sequence | Used for |
|---|
| CCGAATTCATAAGCTGGCGGATTTAATG | pMC1403-ydbK′::lacZ, substrate for gel shift assay |
| CCGGATCCCCGTCAATAGTAATCATATG | Common reverse primer for ydbK′::lacZ reporters substrate for gel shift assay (–562) |
| CACCTGACGTCTAAGAAAC | Sequencing for pMC1403 |
| GGCGAAAGGGGGATGTGCT | Sequencing for pMC1403 |
| CCGAATTCTTCTTCCTCTGATCTTCAAG | for gel shift assay (–270) |
| CCGAATTCTGATGTGGGGGACACAAAAG | pMC1403-ydbK′::lacZ-119, substrate for gel shift assay (–119) |
| CCGAATTCAGCGAAAATGCAGAAGAAAG | pMC1403-ydbK′::lacZ-101, substrate for gel shift assay (–102) |
| CCGAATTCGCGCGCAGCAATTTCGTG | for gel shift assay (–50) |
| CCGAATTCGCGCAATGTAAGGGTGTCAT | for gel shift assay (–20) |
| CCGGATCCGCGTAATGAATGTTTTGATC | Substrate for gel shift assay (–562 to –120) |
| GCGCGAATTCATGTCCCATCAGAAAATTATTCAG | pMal-soxS |
| TAATAAGCTTTTACAGGCGGTGGCGATAATC | pMal-soxS |
| GGTCGTCAGACTGTCGATGAAGCC | Sequencing primer for pMal-c2 |
| AATTCTGAGGATCCTCTAGAGTCGACCTGCAGGCA | pMal-stop |
| AGCTTGCCTGCAGGTCGACTCTAGAGGATCCTCAG | pMal-stop |
| ATGAATTCGCCACACCGCTGCGTTTCGC | pMC1403-soxS′::lacZ |
| ATGGATCCATTTTCTGATGGGACATAAA | pMC1403-soxS′::lacZ |
| ATGAATTCGGGGACACAAAAGCGAAAATGCAGAAGAAAGCC | pMC1403-ydbK′::lacZ-112 |
| ATGAATTCGGGGACACAAAAGCGAAAATATAGAAGAAAGCC | pMC1403-ydbK′::lacZ-#1-1 |
| ATGAATTCGGGGACACCCCAGCGAAAATGCAGAAGAAAGCC | pMC1403-ydbK′::lacZ-#1-2 (#2-2) |
| ATGAATTCGGGGACATTTGAGCGAAAATGCAGAAGAAAGCC | pMC1403-ydbK′::lacZ-#1-3 |
| ATGAATTCGGGGATACAAAAGCGAAAATGCAGAAGAAAGCC | pMC1403-ydbK′::lacZ-#2-1 |
Table 2. Bacterial strains used in this study
Table 3. Plasmids used in this study
| Plasmid name | Features | Reference |
|---|
| pMC1403 | AmpR lacZ’ | Casadaban et al. (1980) |
| pMC1403-ydbK′::lacZ | AmpR ydbK′::lacZ’ | This study |
| pMC1403-ydbK′::lacZ-119 | AmpR ydbK′::lacZ’ | This study |
| pMC1403-ydbK′::lacZ-101 | AmpR ydbK′::lacZ’ | This study |
| pMC1403-ydbK′::lacZ-#1-1 | AmpR ydbK′::lacZ’ | This study |
| pMC1403-ydbK′::lacZ-#1-2 (#2-2) | AmpR ydbK′::lacZ’ | This study |
| pMC1403-ydbK′::lacZ-#1-3 | AmpR ydbK′::lacZ’ | This study |
| pMC1403-ydbK′::lacZ-#2-1 | AmpR ydbK′::lacZ’ | This study |
| pMC1403-soxS′::lacZ | AmpR soxS′::lacZ’ | This study |
| pCP20 | AmpR CmR flp | Cherepanov and Wackernagel (1995) |
| pNT3-ydbK | AmpR ydbK | Saka et al. (2005) |
| pNTR-SD | AmpR | Saka et al. (2005) |
| ASKA soxS | CmR soxS | Kitagawa et al. (2005) |
| pMal-c2 | AmpR malE-lacZα | NEB |
| pMal-c2-soxS | AmpR malE-soxS | This study |
| pMal-c2-stop | AmpR malE | This study |
To remove the Km-resistance gene from the genome, JW3895 (fpr::Km) was transformed with pCP20, which carried FLP recombinase (Cherepanov and Wackernagel, 1995), and clones sensitive to both Km and Ap were selected (fpr mutants without a kanamycin cassette and pCP20). The ydbK::Km construct was then transduced from JW1372 to its fpr mutant strain using the P1 phage (Saka et al., 2005; Kitagawa et al., 2005).
Construction and mutagenesis of gene fusion constructs with lacZ
The upstream sequences of ydbK (–562 to +17, –119 to +17 and –102 to +17) and soxS (–156 to +17) were amplified by PCR using primers for introducing EcoRI and BamHI restriction sites at the end of the product (Table 1). PCR products were purified using a High Pure PCR Cleanup Micro Kit (Roche). pMC1403 and purified PCR products were digested with EcoRI and BamHI, purified by agarose gel electrophoresis and recovered using a UltraClean 15 DNA Purification Kit (MO BIO). The ligation reaction was performed using a DNA Ligation Kit Ver. 2.1 (Takara), and the products were used to transform competent cells of the E. coli strain XL10-GOLD (Table 2). Transformants were selected on LB plates supplemented with Ap. Plasmid DNA was purified from Ap-resistant colonies using a Wizard minicolumn (Promega) and sequenced by a fluorescent dideoxy method (Smith et al., 1986).
Mutagenesis in the ydbK promoter sequence was performed by using primers that contain mutations (Table 1).
Purification of MalE-SoxS protein
The soxS coding sequence was amplified by PCR using primers to introduce EcoRI and HindIII restriction sites at the ends of the product (Table 1). pMal-c2 (New England BioLads) and purified PCR products were digested with EcoRI and HindIII.
Purification of the MalE-SoxS protein was conducted according to the method of Fawcett and Wolf (1994) with a slight modification. Overnight cultures were diluted 100-fold in LB medium supplemented with Ap (50 μg/ml) and glucose (2 mg/ml), and grown at 37℃ until the OD600 reached 0.4–0.5. Isopropyl-β-D-thiogalactopyranoside (IPTG) was added at a final concentration of 0.1 mM and incubation was continued for 4 h. Cells were pelleted by centrifugation at 7,000 rpm at 4℃, washed with PBS and frozen at –20℃ before use. Cells were resuspended in 10 volumes of 1 x buffer A (20 mM HEPES (pH 7.4) containing 1 mM EDTA, 200 mM NaCl, 1 mM dithiothreitol (DTT)) and disrupted by sonication at 4℃. The lysates were centrifuged at 14,000 rpm for 10 min at 4℃ and the supernatant was kept in fresh tubes. The cell pellets were resuspended in 5 volumes of buffer A, disrupted and centrifuged at 14,000 rpm for 10 min at 4℃. The supernatants were mixed and centrifuged at 14,000 rpm at 4℃ for 30 min. The supernatant was subsequently loaded on a 10-ml column packed with amylose resin (New England BioLabs). The column was washed twice with buffer A, and eluted with elution buffer (buffer A containing 20 mM maltose). Fractions were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE). FactorXa (1% w/w, New England BioLads) was added to the fractions exhibiting peak protein content and incubated for 20 h on ice. Then, an equal volume of 2 x buffer A containing 80% glycerol and 2 mM phenylmethylsulfonyl fluoride (PMSF) was added. Samples were stored at –20℃ prior to use. Protein concentrations of undigested samples were determined using a Bradford assay with BSA as the standard.
MalE protein was purified from E. coli cells harboring pMal-stop with same procedures.
Assay for pyruvate oxidoreductase activity
All buffers were freed from oxygen by flushing with nitrogen for at least 2 min immediately before use. E. coli cells were cultured aerobically in LB medium and suspended in extraction buffer (50 mM Tris-HCl (pH 7.5) containing 20% ethylene glycol, 1 mM MgCl2, 0.1 mM thiamine diphosphate (TPP), 10 mM thiosulfate, 50 mM β-mercaptoethanol and 1 mM DTT). Cells were disrupted by sonication at 4℃. The lysates were centrifuged at 14,000 rpm for 30 min at 4℃ and the supernatants (crude extracts) were kept on ice. The reaction mixture (5 ml) contained 100 mM Tris-HCl (pH 7.5), 5 mM MgSO4, 6 mM DTT, 0.1 mM TPP, 0.2 mM CoA, 10 mM pyruvate and 1 mM MV. After oxygen was removed by aspiration for 10 min, the crude extract was added to the reaction mixture. PFOR activity was assayed at room temperature by monitoring the reduction of MV. The increase in absorbance at 578 nm was measured in anaerobic Thunberg tubes (ϕ18 mm).
Assay for sensitivity to MV
E. coli cells were grown in LB medium at 37℃ to a stationary phase, washed and diluted 10-fold with PBS. Sterilized paper strips (0.5 × 12 cm) were put in the cell suspensions, kept at room temperature for 5 min and then placed on LB plates (10.4 × 14.4 cm) containing MV with a linear gradient of concentrations (0–0.8 mM or 0–0.2 mM for the ydbK fpr double mutant). After the paper strips were removed, the plates were incubated at 37℃ for about 18 h, and the zone of growth inhibition was determined to estimate survival.
Assay for aconitase activity
Aconitase activity was measured according to the method described by Skovran and Downs (2000) with a slight modification. Cells were grown in LB medium overnight and diluted 100-fold in 50 ml of fresh LB medium. The cultures were incubated until the OD600 reached 0.5. After treatment with 100 μg/ml of MV for 60 min, the cells were harvested, washed and resuspended in 4 ml of buffer B (150 mM Tris-HCl (pH 8.0) containing 12.5 mM citrate and 1.5 mM NaCl). The cells were lysed by sonication at 4℃ and the lysates were centrifuged at 14,000 rpm for 5 min at 4℃. The extracts were mixed with 20 mM isocitrate and aconitase activity was assayed at room temperature by monitoring the increase in absorbance at 240 nm after addition of the crude extract. Protein concentrations were determined using the Bradford assay with BSA as the standard, and specific activity was calculated as the change in absorbance/min/mg of protein.
β-galactosidase assay
The overnight cultures were diluted 100-fold into fresh LB medium unless otherwise stated. The β-galactosidase activity of whole cell extracts was determined as described by Miller (Miller, 1972). The activity was estimated as units/OD600.
Gel shift assay
DNA fragments were amplified by PCR using the primers listed in Table 1. Reverse primers were end-labeled with [γ-32P]-ATP using T4 polynucleotide kinase. The DNA-binding reaction for the gel shift assay was performed in 30 μl of reaction mixture (24 mM HEPES (pH 7.9), 5 mM Tris, 20 mM NaCl, 50 mM KCl, 1.4 mM EDTA, 0.4 mg/ml BSA, 18% glycerol, 1 mM DTT, 500 μg of calf thymus DNA, 5–10 fmol of substrate duplex oligonucleotide and 0–5 pmol of purified SoxS or 5 pmol purified MalE). The reaction mixture was incubated on ice for 15 min and loaded on prerun native acrylamide gels. Gels were run against TGE buffer (25 mM Tris-HCl (pH 8.3), 190 mM glycine and 100 mM EDTA) at 300 V for 1 min, and then at 125 V for 3 h at 4℃.
RESULTS
The product of the ydbK gene has PFOR activity
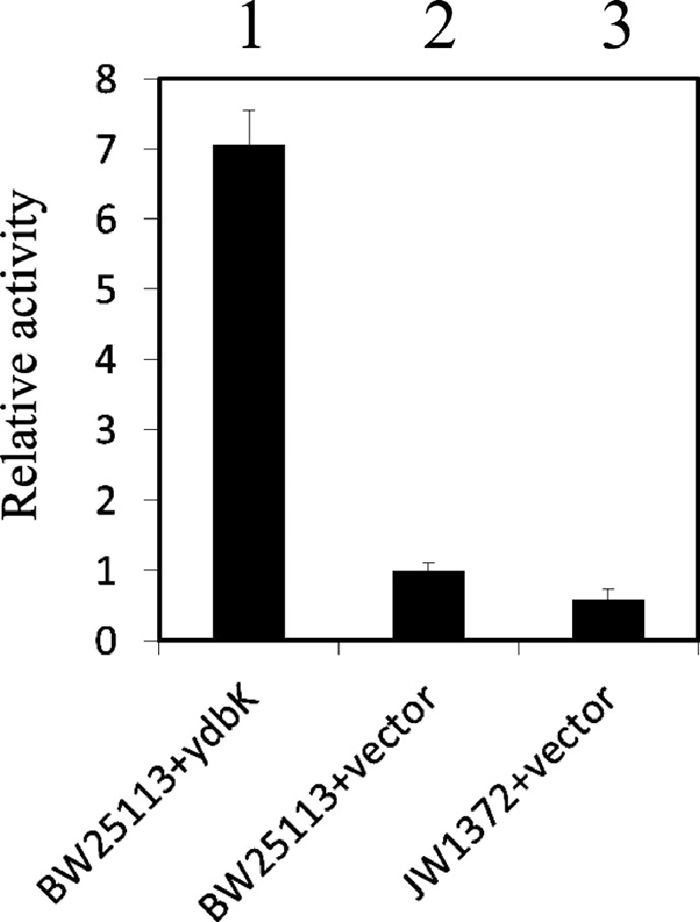
Computational analysis has predicted that E. coli YdbK is PFOR (Serres et al., 2001). Because the PFOR was extremely labile to oxygen, its enzymatic activity and the regulation of its gene expression are not well understood. In this study, we attempted to characterize the enzymatic activity of YdbK in vivo. We measured the PFOR activity in crude extracts prepared from various E. coli cells by monitoring the reduction of MV. MV effectively substitutes for flavodoxin as an electron acceptor in pyruvate oxidation (Kaihovaara et al., 1998). In the extract from the E. coli ydbK mutant, the oxidoreductase activity decreased to about 40% (Fig. 1). On the other hand, a highly elevated level of pyruvate oxidoreductase activity was detected in the crude extract from E. coli cells harboring a YdbK-expressing plasmid (Fig. 1). The PFOR activity increased about 7-fold. These results indicate that the E. coli ydbK gene product is PFOR.
The fpr ydbK double mutant exhibits hypersensitivity to MV
E. coli with mutations in both ydbK and fpr genes displays abnormal sensitivity to MV (Eremina et al., 2010; Bianchi et al., 1995). The fpr gene is also regulated by SoxS and encodes a ferredoxin (flavodoxin)-NADP(H) reductase (FPR) (Krapp et al., 2002). It has been predicted that YdbK also delivers reducing equivalents to flavodoxin (Serres et al., 2001). Hence we constructed a fpr ydbK double mutant and characterized its phenotypes. The double mutant exhibited slow growth in LB medium under aerobic conditions compared with wild-type and single mutant strains, and sometimes aggregated in overnight cultures (data not shown). To assess MV sensitivity, overnight cultures were plated on agar plates containing a linear gradient of MV, and subjected to survival assays. The double mutant was more sensitive to MV than the fpr or ydbK single mutant (Fig. 2, Table 4).

Table 4. Zone of growth inhibition (mm)
| Genotype | Zone of growth inhibition (mm) |
|---|
| Expt. 1 | Expt. 2 | Expt. 3 | Mean | S.D. |
|---|
| wild-type | 33 | 20 | 23 | 25.3 | 6.8 |
| ydbK | 60 | 55 | 50 | 55.0 | 5.0 |
| fpr | 100 | 94 | 94 | 96.0 | 3.5 |
| ydbK fpr (clone 1) | 123 | 103 | 120 | 115.3 | 10.8 |
| ydbK fpr (clone 2) | 117 | 126 | 107 | 116.7 | 9.5 |
E. coli cells were grown at 37℃ to a stationary phase, washed and diluted 10-fold with PBS. Sterilized paper strips (0.5 × 12 cm) were put in the cell suspensions and then placed on LB plates (10.4 × 14.4 cm) containing MV with a linear gradient of concentrations, followed by incubation at 37℃ for about 18 h. The zone of growth inhibition was determined to estimate survival.
E.coli has two aconitases, AcnA and AcnB (Gruer and Guest, 1994). AcnB is the major aconitase and acnB gene was activated early in the exponential phase (Gruer et al., 1997). The active center of aconitase B, which contains an 4Fe-4S cluster, is labile to superoxide, therefore, aconitase activity is considered to be an indicator of oxidative stress in E. coli. To elucidate the in vivo functions of YdbK under oxidative stress, we measured the aconitase activity in wild-type and ydbK mutant strains incubated with or without 100 μg/ml MV for 1 h. The aconitase activity was decreased by MV in both strains, and the decrease was greater in MV-treated ydbK mutant (54.9%) than MV-treated wild-type strain (38.3%) (Fig. 3).
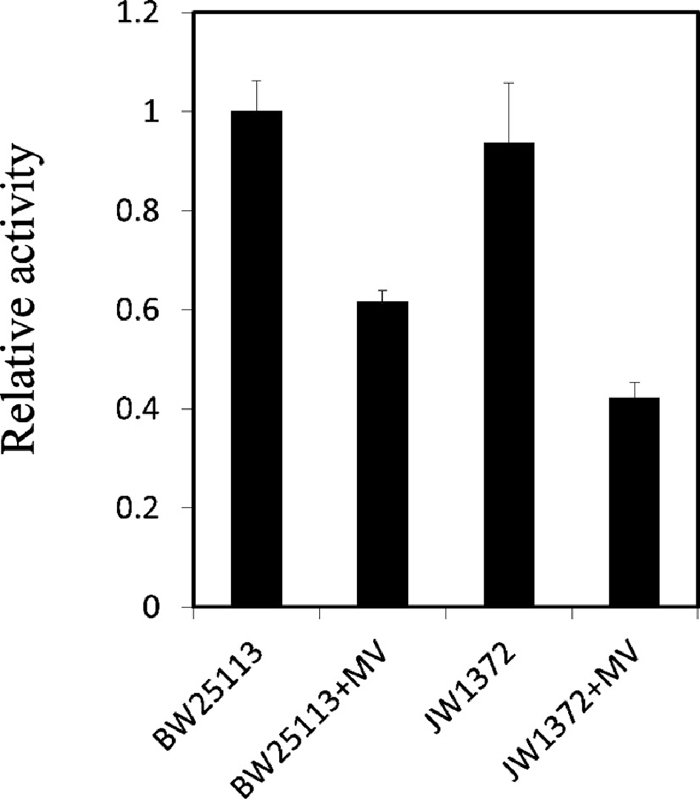
AcnA is an oxygen-resistant isozyme and induced by superoxide in a SoxS-dependent manner (Gardner and Fridovich, 1991; Varghese et al., 2003). However, the induction of acnA gene is slower and the induction level is lower than other superoxide-inducible genes (Lu et al., 2005). These facts suggest that MV-induced acnA expression did not completely rescue the loss of AcnB activity caused by MV and that the decrease in the AcnB activity was greater than the induction of AcnA activity after treatment with MV.
SoxS-dependent induction of ydbK’::lacZ fusion gene
To investigate the regulatory mechanisms of expression of ydbK gene, we used a gene fusion construct with lacZ gene as a reporter. The resultant pMC1403/ydbK’::lacZ plasmid contained the 562-bp upstream region and the first several N-terminal amino acids of the YdbK protein, fused to the lacZ gene of the plasmid vector in frame. We introduced the plasmid into E. coli GC4468 and GC4468soxS and assayed the β-galactosidase activity in these cells 1 h after treatment with various concentrations of MV. The expression of the ydbK’::lacZ fusion gene was induced by MV, which was eliminated by the soxS gene disruption (Fig. 4A). Furthermore, SoxS overexpression by incubating E. coli cells with 0.4 mM IPTG for up to 90 min enhanced the expression of ydbK’::lacZ without MV (Fig. 4B). Recent studies showed that SoxRS regulon genes are also induced in response to hydrogen peroxide (H2O2) (Semchyshyn et al., 2005). In this study, however, we observed that H2O2 did not induce the expression of ydbK’::lacZ fusion gene (data not shown). Furthermore, we also obtained the results that the response of ydbK’::lacZ to MV was significantly eliminated in E.coli by disruption of soxR gene (data not shown). These results indicate that transcriptional activation of the ydbK gene is markedly dependent on the function of SoxS. It was also found that the induction of ydbK’::lacZ occurred after a lag time (Fig. 4B).
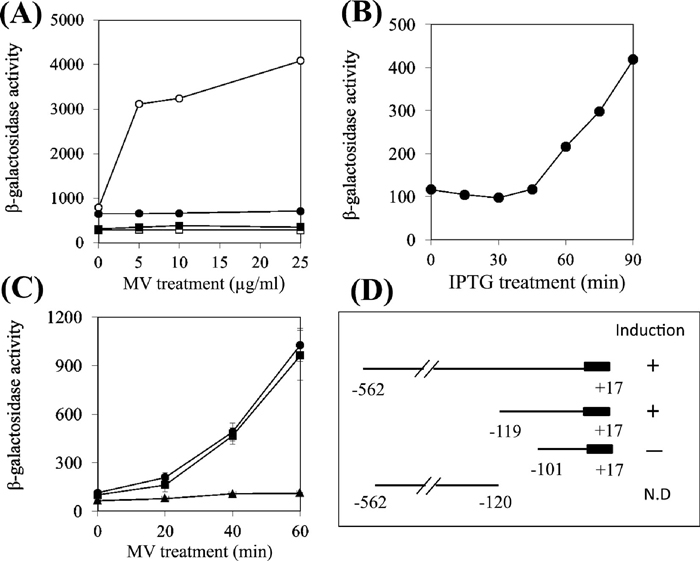
Next, to identify the region critical for the MV response, we generated a series of 5’ deletions of the upstream region of the ydbK gene fused to lacZ gene. Cultures were treated with 10 μg/ml (32 μM) of MV for up to 1 h. The lacZ reporter could responded to MV treatment when fused to the 562-bp and 119-bp upstream region of the ydbK gene, but not when fused to the 101-bp region (Fig. 4, C and D), indicating that the sequence between –119 and –102 is critical for the MV response. The expression of ydbK’::lacZ increased after a lag time of more than 20 min of exposure to MV (Fig. 4C).
Binding of SoxS to the ydbK promoter region in vitro
Members of the AraC family share low solubility (Egan and Schleif, 1994; Rodgers and Schleif, 2009). MalE is a very effective solubilizing fusion partner (Kapust and Waugh, 1999). MalE-SoxS was previously shown to be able to recognize the SoxS-binding sequence (Fawcett and Wolf, 1994). Hence, we purified the MalE-SoxS protein (Fig. 5A) and confirmed by SDS-PAGE. Fig. 5A indicates that we have purified MalE-SoxS protein successfully, and the results of in vitro assay were not due to other contaminated proteins.
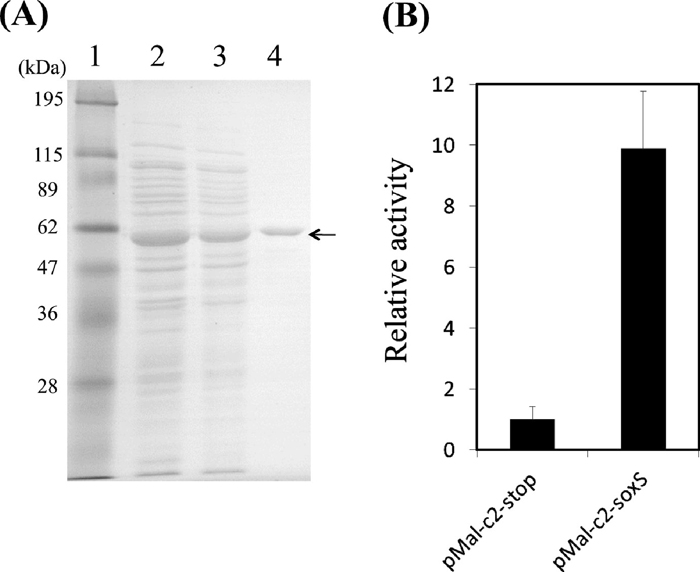
Prior to the in vitro analysis, we investigated whether overexpression of MalE-SoxS by incubation with IPTG induces expression of the ydbK gene in vivo. We introduced pMal-c2-soxS or pMal-c2-stop into the E. coli soxS mutant and measured YdbK activity in the cell extract. As shown in Fig. 5B, we could detect approximately 10-fold higher activity of PFOR in E. coli overexpressing MalE-SoxS than in control E. coli cells. Furthermore, a high level of the 128-kDa protein was also observed in MalE-SoxS-overexpressing E. coli cells by SDS-PAGE (data not shown). These results indicate that MalE-SoxS can also induce the expression of ydbK in vivo.
However, the MalE-SoxS showed a weak binding activity to the promotor of ydbK gene in vitro. So, we digested purified MalE-SoxS with Factor Xa (1% w/w) and used the mixture in the presence of PMSF. Purified MalE was used as a negative control.
A gel shift assay was conducted with 32P-labeled duplex oligonucleotides prepared by PCR using primers listed in Table 1. As shown in Fig. 6A, SoxS formed a complex with DNA fragments that contain the sequence from –562 to +17 (fragment I), but such binding was not observed with MalE. With fragment I, two complexes with different sizes were observed as the amount of SoxS increased. Similar binding properties of SoxS have been also reported in micF, zwf, nfo and sodA (Li and Demple, 1994). Oligomerization of SoxS is unlikely because it binds as a monomer (Jair et al., 1996), suggesting that there are multiple SoxS-binding sites in the ydbK promoter. This is consistent with the fact that SoxS could also form a complex with a DNA fragment containing the sequence between –562 and –120 (data not shown). However, because the deletion of this secondary binding site did not influence the induction of ydbK’::lacZ fusion by MV (Fig. 4C), this site is not involved in the MV response.
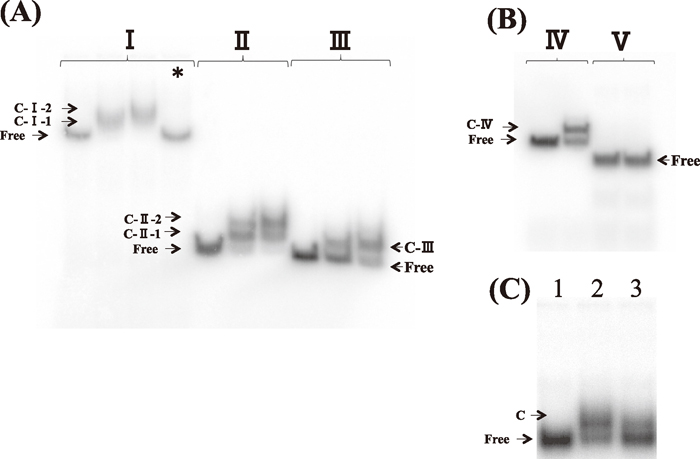
When we tested DNA fragments that contained the sequence from –119 to +17 (fragment II), SoxS formed two complexs with different size, while only single complex was observed with DNA fragments that contained the sequence from –101 to +17 (fragment III) (Fig. 6A). Importantly, SoxS also formed complex with DNA fragments that contained the sequence from –50 to +17 (Fig. 6B, fragment IV). Since –35 hexamer that is recognized by RNAP is known to overlap or lie the downstream of soxbox, deletion of sequence from –119 to –102 did not remove –35 hexamer of the downstream binding site.
Since SoxS did not form complex with DNA fragments that contained the sequence from –20 to +17 (Fig. 6B, fragment V), indicating that at least part of downstream binding site is overlapped at the region from –49 to –21.
To examine a possibility of contamination of other proteins that bind ydbK promoter sequence, we performed a competition assay. We used 32P-labeled zwf upstream sequence (–300 to +17) as a probe that contains a SoxS-binding site (Fawcett and Wolf, 1994), and added unlabeled ydbK upstream sequence with similar length (–270 to +17) as a competitor. When 60 ng of ydbK fragment was added, significant inhibitory effect was observed (Fig. 6C). All samples used contained 500 ng of calf thymus DNA to stabilize SoxS protein. More than 1,200 ng of calf thymus DNA was required to obtain the same extent of inhibitory effect. These results indicate that SoxS specifically binds to ydbK promoter sequence.
Mutation analysis of the ydbK promoter
Previously reported consensus sequences (“Soxbox”) for SoxS-binding have three characteristics (Griffith and Wolf, 2001); (1) an invariant A at position 1, (2) two recognition elements (RE1 and RE2), to which two HTH motifs of SoxS bind, and (3) an AT-rich spacer between the two REs (Fig. 7). Promoters can be divided in two classes based on the position and orientation of the soxbox in relation to the –35 hexamer.

In the region of the upstream sequence of ydbK, there were two putative SoxS -binding sites (Fig. 7). In the non-template strand, which is in the reverse orientation, there is an element (soxbox 1) that is similar to the Soxbox of the rob promoter (Schneiders and Levy, 2006). This element shares a 7nt stretch that includes the RE1 element (GCAT). The same 7nt stretch is also found in the Soxbox of the nfnB promoter (Barbosa and Levy, 2002). This putative element did not have the invariant A of rob and nfnB. There is no similarity with the consensus sequence (CAAA) in the RE2 element. Instead of a string of A’s, this putative binding site has a T-rich sequence on its 3’ side. In the template strand (in the forward orientation), we could detect a 20-bp element (soxbox 2) that shared 12/17 specified nucleotides with the consensus sequence. This element also lacked the invariant A and the first nucleotide of the RE1 element was A instead of G. This stretch (ACAC) was also found in the soxbox of the inaA promoter (Martin and Rosner, 2011).
To clarify which element is the genuine binding site, we introduced several mutations and investigated the effect on the expression of ydbK’::lacZ fusion and on the binding of MalE-SoxS. The results are shown in Table 5. The oligonucleotides #1-1 and #2-1 contained a mutation in the RE1 element of soxbox 1 and soxbox 2, respectively. #1-2 carried a mutation in the RE2 element of soxbox 1, and #2-2, which had the same sequence as #1-2, carried a mutation in the AT-rich spacer of soxbox 2. In #1-3, the RE2 element of soxbox 1 was modified to generate the consensus sequence CAAA. In the case of soxbox 2, mutation of RE2 was constrained because it probably overlaps with the –35 hexamer.
Table 5. Mutagenesis of soxboxes
| Feature | Sequence | Induction (%) | Binding of MalE-SoxS |
|---|
| soxbox 1 | non-template strand, –107 to –88 | TCTGCATTTTCGCTTTTGTG | 100.0 | + |
| #1-1 | mutation in RE1 | TCTATATTTTCGCTTTTGTG | 93.6 | + |
| #1-2 | #1-2 mutation in RE2 | TCTGCATTTTCGCTGGGGTG | 11.0 | — |
| #1-3 | RE2→consensus | TCTGCATTTTCGCTCAAATG | 15.3 | — |
| soxbox 2 | template strand, –111 to –92 | GGGACACAAAAGCGAAAATG | 100.0 | + |
| #2-1 | mutation in RE1 | GGGATACAAAAGCGAAAATG | 12.7 | — |
| #2-2 | mutation in spacer | GGGACACCCCAGCGAAAATG | 15.3 | — |
Various E. coli cells were treated with MV and the β-galactosidase activity of whole cell extract was determined as described by Miller (1972). The DNA-binding reaction for the gel shift assay was performed as described in EXPERIMENTAL PROCEDURES. Samples were loaded on prerun 6% native acrylamide gels. Gels were run against TGE buffer (25 mM Tris-HCl (pH 8.3), 190 mM glycine and 100 mM EDTA) at 300 V for 1 min, and then at 125 V for 3 h at 4℃.
In the case of #1-2 and #1-3, ydbK’::lacZ fusions were not induced by MV, and MalE-SoxS did not bind to the DNA fragments. On the other hand, the expression of lacZ and the binding of MalE-SoxS were not affected by the mutations introduced in #1-1. These results indicate that at least 3 T’s located in the RE2 of soxbox 1 are important but soxbox 1 was not a genuine binding site.
Because #2-2 had the same sequence as #1-2 but was located in the opposite strand, the 3 A’s located in the spacer region must be critical. Moreover, a single mutation in the RE1 of soxbox 2 (ACAC to ATAC) was deleterious, indicating that soxbox 2 was the genuine binding site.
We could not find the downstream soxbox, and introduction of mutations was constrained because of its influence on other important elements such as Shine-Dalgano sequence.
Effects of YdbK levels on the induction of SoxS
The results described above suggest that the cellular redox state is disturbed in the E. coli ydbK mutant. Hence, we investigated whether the induction of SoxS was influenced by the level of YdbK. A soxS’::lacZ fusion gene was constructed on a plasmid and introduced into a ydbK-proficient or deficient E. coli strain. In the presence of 10 μg/ml MV, the expression level of soxS’::lacZ was not significantly affected by a single mutation in the ydbK gene (data not shown). Furthermore, previous studies showed that a single mutation of fpr did not affect the induction of the SoxRS regulon (Krapp et al., 2002). The E. coli ydbK fpr double mutant was highly sensitive to MV (Table 4), indicating their mutual backup interactions. Therefore, we measured the expression of soxS’::lacZ in the double mutant. Overnight culture was diluted appropriately and grown until the OD600 reached 0.2, followed by treatment with 2 μg/ml (6.4 μM) of MV for up to 1 h. Compared with the wild-type strain, the expression of soxS’::lacZ was remarkably higher in the mutant strain (Fig. 8A). Even in the absence of MV, significantly higher expression was observed in the mutant strain, indicating that disruption of both ydbK and fpr was enough to disturb the cellular redox state in E. coli under aerobic conditions.
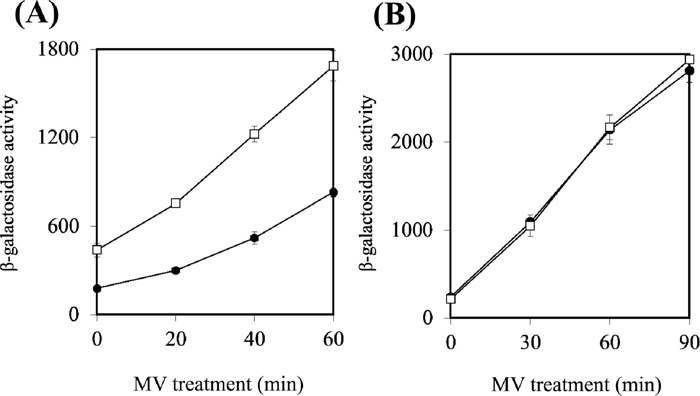
We next investigated the effect of YdbK overexpression. pNT3-ydbK or the supporting vector was introduced into E. coli TN521 carrying soxS’::lacZ and a intact soxR gene in λ lysogen. The overnight culture was diluted appropriately and grown until the OD600 reached 0.4–0.5. IPTG was then added at a final concentration of 0.5 mM and further incubated for 3 h. The cells were treated with 10 μg/ml (32 μM) of MV for up to 90 min. Although a high level of production of the 128-kDa protein was detected by SDS-PAGE (data not shown), the expression of soxS’::lacZ was not changed by overexpression of YdbK (Fig. 8B).
DISCUSSION
YdbK has a PFOR activity
Serres et al. (2001) has predicted that the E. coli YdbK protein is PFOR. However, it’s enzymatic activity still unclear. In this study, we characterized the enzymatic activity of YdbK in vitro and in vivo. Purification of YdbK was so difficult because of its extreme sensitivity to oxygen. Furthermore, in the extract from the E. coli ydbK mutant, the oxidoreductase activity decreased to only 40% compared with wild-type cells (Fig. 1). The residual activity is probably due to other pyruvate-oxidizing enzymes such as pyruvate formate-lyase and pyruvate dehydrogenase complex (Blaschkowski et al., 1982; Knappe et al., 1984). Hence, we attempted to characterize the enzymatic activity in crude extract prepared from E. coli harboring a YdbK-overexpressing plasmid. We detected a highly elevated level of PFOR activity in crude extract from YdbK-overexpressing E. coli (Fig. 1). It is therefore concluded that the E. coli ydbK gene encodes PFOR.
ydbK is induced by superoxide and directly regulated by SoxS
The ydbK gene was induced by MV in a SoxS-dependent manner under aerobic conditions (Fig. 4). The expression of ydbK (previously named soi-9) was induced by MV, menadione bisulfite and exposure to high pressure oxygen (Mito et al., 1993), but not by H2O2, indicating that it is a cellular response to superoxide radical. In Salmonella enterica, a ydbK-like gene was also shown to be induced by MV in a SoxS-dependent manner and the mutant of this gene was sensitive to MV (Pomposiello and Demple, 2000). In addition, flavodoxin A and flavodoxin B, the redox partners of YdbK, are members of the regulon (Gaudu and Weiss, 2000).
MarA and Rob also belong to the AraC/XylS family and are known to activate transcription of a common set of genes (Martin and Rosner, 2011). Salicylate and 2,2’-dipyridyl specifically activate the Mar- and Rob-regulon, respectively (Pomposiello and Demple, 2001; Martin and Rosner, 2004; Rosner et al., 2002). However, in the E. coli soxS mutant strain, salicylate (5 mM) and 2,2’-dipyridyl (5 mM) did not induce ydbK’::lacZ expression significantly (data not shown), indicating that MarA and Rob are not involved in ydbK expression. YdbK’s induction seems to be specific to oxidative stress.
In this study, it is concluded that the sequence between –119 and –102 upstream of the ydbK promotor is critical for the MV response. The argument was derived from the following results; (1) The lacZ reporter could respond to MV treatment when fused to the 562-bp and 119-bp upstream region of the ydbK gene, but not when fused to the 101-bp region (Fig. 4C). (2) SoxS formed two complexes with DNA fragment that contain the sequence between –119 to +17, but only one complex was formed when the fragment containing the sequence from –101 and +17 was used.
The expression of ydbK was induced after a lag period
It has been shown that the extent of SoxR activation is greater in FPR-overexpressing cells, because FPR consumes NADPH, an important reducing source, and SoxS induces FPR only after a lag period of several minutes to avoid this problem (Krapp et al., 2002). ydbK’::lacZ was also expressed after a lag period (Fig. 4). Both YdbK and FPR transfer an electron to flavodoxin. These results indicate that, in addition to loss of NADPH and oxidative deactivation of YdbK, excess reduction of flavodoxin in the presence of redox-cycling agents may need to be avoided.
Protection against oxidative stress
The fpr ydbK double mutant was more sensitive to MV than the fpr or ydbK single mutant (Table 4). These results indicate that FPR and YdbK play antioxidative roles in independent but related pathways. FPR functions to restore reducing equivalents and/or reactivates damaged enzymes (Fig. 9). Giró et al. (2006) reported that FPR-deficient mutants were still able to reactivate Fe-S clusters and suggested the existence of back-up routes. Because aconitase activity was reduced in YdbK-deficient cells (Fig. 3), YdbK may function in this back-up route (Fig. 9).
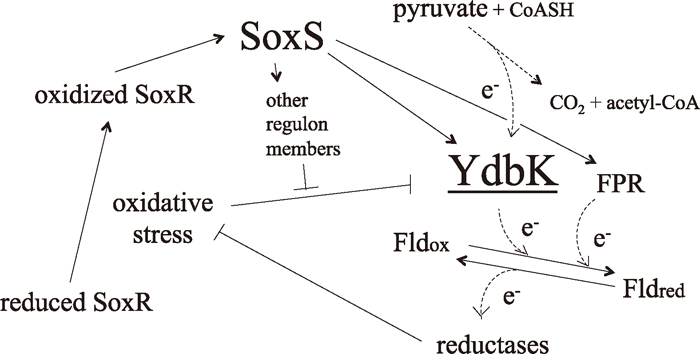
From the view point of iron metabolism, it seems reasonable that oxidative stress induces YdbK expression. It has been shown that superoxide liberated iron from storage proteins or iron-sulfur clusters (Keyer and Imlay, 1996; McCormick et al., 1998). Free iron easily reacts with H2O2 and generates hydroxyl radicals that damage cellular components such as DNA. SoxRS regulon also includes Fur, the global repressor of iron uptake (Zheng et al., 1999). It is known that, under iron-deficient conditions, ferredoxin-NADP+ oxidoreductase is reduced by flavodoxin instead of by ferredoxin, an iron- containing electron donor (Osborne et al., 1991). So, YdbK, a redox partner of flavodoxin, may be necessary to reserve reducing equivalents under such conditions.
ACKNOWLEDGMENT
We thank Dr. K. Hashiguchi for construction of the pMal-stop plasmid. We also thank Drs. B. Demple and T. Nunoshiba and the National BioResource Project (NIG, Japan) for kindly supplying the E. coli strains and plasmids used in this study. This work was financially supported in part by Grants-in-aid for Scientific Research (#24510071) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to Qiu-Mei Zhang-Akiyama) and the Global Center of Excellence Program “Formation of a Strategic Base for Biodiversity and Evolutionary Research (A06): from Genome to Ecosystem”. We are grateful to the Shiseido Female Research Science Grant for supporting Qiu-Mei Zhang-Akiyama.
REFERENCES
- Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., Datsenko, K., Tomita, M., Wanner, B., and Mori, H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008.
- Barbosa, T. M., and Levy, S. B. (2002) Activation of the Escherichia coli nfnB gene by MarA through a highly divergent marbox in a class II promoter. Mol. Microbiol. 45, 191–202.
- Bianchi, V., Haggard-Ljungquist, E., Pontis, E., and Reichard, P. (1995) Interruption of the ferredoxin (flavodoxin) NADP+ oxidoreductase gene of Escherichia coli does not affect anaerobic growth but increases sensitivity to paraquat. J. Bacteriol. 177, 4528–4531.
- Blaschkowski, H. P., Neuer, G., Ludwig-Festl, M., and Knappe, J. (1982) Routes of flavodoxin and ferredoxin reduction in Escherichia coli. CoA-acylating pyruvate: flavodoxin and NADPH: flavodoxin oxidoreductases participating in the activation of pyruvate formate-lyase. Eur. J. Biochem. 123, 563–569.
- Carlioz, A., and Touati, D. (1986) Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? EMBO J. 5, 623–630.
- Casadaban, M. J., Chou, J., and Cohen, S. N. (1980) In vitro gene fusions that join an enzymatically active beta-galactosidase segment to amino-terminal fragments of exogenous proteins: Escherichia coli plasmid vectors for the detection and cloning of translational initiation signals. J. Bacteriol. 143, 971–980.
- Cherepanov, P. P., and Wackernagel, W. (1995) Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14.
- Cui, H., Kong, Y., and Zhang, H. (2012) Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct., Doi:10.1155/2012/646354.
- Datsenko, K. A., and Wanner, B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645.
- Egan, S. M., and Schleif, R. F. (1994) DNA-dependent renaturation of an insoluble DNA binding protein. Identification of the RhaS binding site at rhaBAD. J. Mol. Biol. 243, 821–829.
- Eremina, N. S., Yampolskaya, T. A., Altman, I. B., Mashko, S. V., and Stoynova, N. V. (2010) Overexpression of ydbK-encoding putative pyruvate synthase improves L-valine production and aerobic growth on ethanol media by an Escherichia coli strain carrying an oxygen-resistant alcohol dehydrogenase. J. Microbial Biochem. Technol. 2, 77–83.
- Fabrega, A., Rosner, J. L., Martin, R. G., Sole, M., and Vila, J. (2012) SoxS-dependent coregulation of ompN and ydbK in a multidrug-resistant Escherichia coli strain. FEMS Microbiol. Lett. 332, 61–67.
- Fawcett, W. P., and Wolf, R. E., Jr. (1994) Purification of a MalE-SoxS fusion protein and identification of the control sites of Escherichia coli superoxide-inducible genes. Mol. Microbiol. 14, 669–679.
- Gardner, P. R., and Fridovich, I. (1991) Superoxide sensitivity of the Escherichia coli aconitase. J. Biol. Chem. 266, 19328–19333.
- Gaudu, P., and Weiss, B. (1996) SoxR, a [2Fe-2S] transcription factor, is active only in its oxidized form. Proc. Natl. Acad. Sci. USA 93, 10094–10098.
- Gaudu, P., and Weiss, B. (2000) Flavodoxin mutants of Escherichia coli K-12. J. Bacteriol. 182, 1788–1793.
- Giró, M., Carrillo, N., and Krapp, A. R. (2006) Glucose-6-phosphate dehydrogenase and ferredoxin-NADP(H) reductase contribute to damage repair during the soxRS response of Escherichia coli. Microbiology 152, 1119–1128.
- Griffith, K. L., and Wolf, R. E., Jr. (2001) Systematic mutagenesis of the DNA binding sites for SoxS in the Escherichia coli zwf and fpr promoters: identifying nucleotides required for DNA binding and transcription activation. Mol. Microbiol. 40, 1141–1154.
- Griffith, K. L., Shah, I. M., and Wolf, R. E., Jr. (2004) Proteolytic degradation of Escherichia coli transcription activators SoxS and MarA as the mechanism for reversing the induction of the superoxide (SoxRS) and multiple antibiotic resistance (Mar) regulons. Mol. Microbiol. 51, 1801–1816.
- Gruer, M. J., and Guest, J. R. (1994) Two genetically-distinct and differentially-regulated aconitases (AcnA and AcnB) in Escherichia coli. Microbiology 140, 2531–2541.
- Gruer, M. J., Bradbury, A. J., and Guest, J. R. (1997) Construction and properties of aconitase mutants of Escherichia coli. Microbiology 143, 1837–1846.
- Gu, M., and Imlay, J. A. (2011) The SoxRS response of Escherichia coli is directly activated by redox-cycling drugs rather than by superoxide. Mol. Microbiol. 79, 1136–1150.
- Imlay, J. A. (2008) Cellular defenses against superoxide and hydrogen peroxide. Annu. Rev. Biochem. 77, 755–776.
- Jair, K. W., Fawcett, W. P., Fujita, N., Ishihama, A., and Wolf, R. E., Jr. (1996) Ambidextrous transcriptional activation by SoxS: requirement for the C-terminal domain of the RNA polymerase alpha subunit in a subset of Escherichia coli superoxide-inducible genes. Mol. Microbiol. 19, 307–317.
- Kaihovaara, P., Hook-Nikanne, J., Uusi-Oukari, M., Kosunen, T. U., and Salaspuro, M. (1998) Flavodoxin-dependent pyruvate oxidation, acetate production and metronidazole reduction by Helicobacter pylori. J. Antimicrob. Chemother. 41, 171–177.
- Kapust, R. B., and Waugh, D. S. (1999) Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 8, 1668–1674.
- Keyer, K., and Imlay, J. A. (1996) Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. USA 93, 13635–13640.
- Kitagawa, M., Ara, T., Arifuzzaman, M., Ioka-Nakamichi, T., Inamoto, E., Toyonaga, H., and Mori, H. (2005) Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12, 291–299.
- Klaunig, J. E., Wang, Z., Pu, X., and Zhou, S. (2011) Oxidative stress and oxidative damage in chemical carcinogenesis. Toxicol. Appl. Pharmacol. 254, 86–99.
- Knappe, J., Neugebauer, F. A., Blaschkowski, H. P., and Ganzler, M. (1984) Post-translational activation introduces a free radical into pyruvate formate-lyase. Proc. Natl. Acad. Sci. USA 81, 1332–1335.
- Kobayashi, K., and Tagawa, S. (1999) Isolation of reductase for SoxR that governs an oxidative response regulon from Escherichia coli. FEBS Lett. 451, 227–230.
- Koo, M. S., Lee, J. H., Rah, S. Y., Yeo, W. S., Lee, J. W., Lee, K. L., Koh, Y. S., Kang, S. O., and Roe, J. H. (2003) A reducing system of the superoxide sensor SoxR in Escherichia coli. EMBO J. 22, 2614–2622.
- Krapp, A. R., Rodriguez, R. E., Poli, H. O., Paladini, D. H., Palatnik, J. F., and Carrillo, N. (2002) The flavoenzyme ferredoxin (flavodoxin)-NADP(H) reductase modulates NADP(H) homeostasis during the soxRS response of Escherichia coli. J. Bacteriol. 184, 1474–1480.
- Kunow, J., Linder, D., and Thauer, R. K. (1995) Pyruvate: ferredoxin oxidoreductase from the sulfate-reducing Archaeoglobus fulgidus: molecular composition, catalytic properties, and sequence alignments. Arch. Microbiol. 163, 21–28.
- Li, Z., and Demple, B. (1994) SoxS, an activator of superoxide stress genes in Escherichia coli. Purification and interaction with DNA. J. Biol. Chem. 269, 18371–18377.
- Lu, C., Albano, C. R., Bentley, W. E., and Rao, G. (2005) Quantitative and kinetic study of oxidative stress regulons using green fluorescent protein. Biotechnol Bioeng. 89, 574–587.
- Lushchak, V. I. (2011) Adaptive response to oxidative stress: Bacteria, fungi, plants and animals. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 153, 175–190.
- Martin, R. G., and Rosner, J. L. (2002) Genomics of the marA/soxS/rob regulon of Escherichia coli: identification of directly activated promoters by application of molecular genetics and informatics to microarray data. Mol. Microbiol. 44, 1611–1624.
- Martin, R. G., and Rosner, J. L. (2004) Transcriptional and translational regulation of the marRAB multiple antibiotic resistance operon in Escherichia coli. Mol. Microbiol. 53, 183–191.
- Martin, R. G., and Rosner, J. L. (2011) Promoter discrimination at class I MarA regulon promoters mediated by glutamic acid 89 of the MarA transcriptional activator of Escherichia coli. J. Bacteriol. 193, 506–515.
- McCormick, M. L., Buettner, G. R., and Britigan, B. E. (1998) Endogenous superoxide dismutase levels regulate iron-dependent hydroxyl radical formation in Escherichia coli exposed to hydrogen peroxide. J. Bacteriol. 180, 622–625
- Miller, J. H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N. Y.
- Mito, S., Zhang, Q. M., and Yonei, S. (1993) Isolation and characterization of Escherichia coli strains containing new gene fusions (soi::lacZ) inducible by superoxide radicals. J. Bacteriol. 175, 2645–2651.
- Nunoshiba, T., and Demple, B. (1993) Potent intracellular oxidative stress exerted by the carcinogen 4-nitroquinoline-N-oxide. Cancer Res. 53, 3250–3252.
- Nunoshiba, T., Hidalgo, E., Li, Z., and Demple, B. (1993) Negative autoregulation by the Escherichia coli SoxS protein. J. Bacteriol 175, 7492–7494.
- Osborne, C., Chen, L. M., and Matthews, R. G. (1991) Isolation, cloning, mapping, and nucleotide sequencing of the gene encoding flavodoxin in Escherichia coli. J. Bacteriol. 173, 1729–1737.
- Pomposiello, P. J., and Demple, B. (2000) Identification of SoxS-regulated genes in Salmonella enterica serovar typhimurium. J. Bacteriol. 182, 23–29.
- Pomposiello, P. J., and Demple, B. (2001) Redox-operated genetic switches: the SoxR and OxyR transcription factors. Trends Biotechnol. 19, 109–114.
- Pomposiello, P. J., Bennik, M. H., and Demple, B. (2001) Genome-wide transcriptional profiling of the Escherichia coli responses to superoxide stress and sodium salicylate. J. Bacteriol. 183, 3890–3902.
- Rodgers, M., and Schleif, R. (2009) Solution structure of the DNA binding domain of AraC protein. Proteins 77, 202–208.
- Rosner, J. L., Dangi, B., Gronenborn, A. M., and Martin, R. G. (2002) Posttranscriptional activation of the transcriptional activator Rob by dipyridyl in Escherichia coli. J. Bacteriol. 184, 1407–1416.
- Saka, K., Tadenuma, M., Nakade, S., Tanaka, N., Sugawara, H., Nishikawa, K., Ichiyoshi, N., Kitagawa, M., Mori, H., Ogasawara, N., and Nishimura, A. (2005) A complete set of Escherichia coli open reading frames in mobile plasmids facilitating genetic studies. DNA Res. 12, 63–68.
- Schneiders, T., and Levy, S. B. (2006) MarA-mediated transcriptional repression of the rob promoter. J. Biol. Chem. 281, 10049–10055.
- Semchyshyn, H., Bagnyukova, T., and Lushchak, V. (2005) Involvement of soxRS regulon in response of Escherichia coli to oxidative stress induced by hydrogen peroxide. Biochemistry (Mosc). 70, 1238–1244.
- Serres, M. H., Gopal, S., Nahum, L. A., Liang, P., Gaasterland, T., and Riley, M. (2001) A functional update of the Escherichia coli K-12 genome. Genome Biol. 2, research0035. I-0035.7.
- Skovran, E., and Downs, D. M. (2000) Metabolic defects caused by mutations in the isc gene cluster in Salmonella enterica serovar typhimurium: implications for thiamine synthesis. J. Bacteriol. 182, 3896–3903.
- Smith, L. M., Sanders, J. Z., Kaiser, R. J., Hughes, P., Dodd, C., Connell, C. R., Heiner, C., Kent, S. B., and Hood, L. E. (1986) Fluorescence detection in automated DNA sequence analysis. Nature 321, 674–679.
- Tsaneva, I. R., and Weiss, B. (1990) SoxR, a locus governing a superoxide response regulon in Escherichia coli K-12. J. Bacteriol. 172, 4197–4205.
- Varghese, S., Tang, Y., and Imlay, J. A. (2003) Contrasting sensitivities of Escherichia coli aconitases A and B to oxidation and iron depletion. J. Bacteriol. 185, 221–230.
- Wahl, R. C., and Orme-Johnson, W. H. (1987) Clostridial pyruvate oxidoreductase and the pyruvate-oxidizing enzyme specific to nitrogen fixation in Klebsiella pneumoniae are similar enzymes. J. Biol. Chem. 262, 10489–10496.
- Williams, K., Lowe, P. N., and Leadlay, P. F. (1987) Purification and characterization of pyruvate: ferredoxin oxidoreductase from the anaerobic protozoon Trichomonas vaginalis. Biochem. J. 246, 529–536.
- Wu, J., and Weiss, B. (1991) Two divergently transcribed genes, soxR and soxS, control a superoxide response regulon of Escherichia coli. J. Bacteriol. 173, 2864–2871.
- Zheng, M., Doan, B., Schneider, T. D., and Storz, G. (1999) OxyR and SoxRS regulation of fur. J. Bacteriol. 181, 4639–4643.