Full papers
WCOACH: Protein complex prediction in weighted PPI networks
2015 Volume 90 Issue 5 Pages 317-324


Details
2015 Volume 90 Issue 5 Pages 317-324
Protein complexes are aggregates of protein molecules that play important roles in biological processes. Detecting protein complexes from protein-protein interaction (PPI) networks is one of the most challenging problems in computational biology, and many computational methods have been developed to solve this problem. Generally, these methods yield high false positive rates. In this article, a semantic similarity measure between proteins, based on Gene Ontology (GO) structure, is applied to weigh PPI networks. Consequently, one of the well-known methods, COACH, has been improved to be compatible with weighted PPI networks for protein complex detection. The new method, WCOACH, is compared to the COACH, ClusterOne, IPCA, CORE, OH-PIN, HC-PIN and MCODE methods on several PPI networks such as DIP, Krogan, Gavin 2002 and MIPS. WCOACH can be applied as a fast and high-performance algorithm to predict protein complexes in weighted PPI networks. All data and programs are freely available at http://bioinformatics.aut.ac.ir/wcoach.
Proteins are very important molecules that physically interact with each other during their participation in biological processes (Srihari and Leong, 2013). Many experimental techniques such as yeast two-hybrid assays (Ito et al., 2001) and mass spectrometry (Gavin et al., 2002) have been developed to discover pairwise protein-protein interactions and construct protein-protein interaction (PPI) networks. Although these methods identify if there is an interaction between two proteins, they cannot detect interactions involving more than two protein partners (Li et al., 2010; Moschopoulos et al., 2011). To understand a biological function, it is useful to determine protein complexes, groups of interacting proteins that participate in the process (Pizzuti and Rombo, 2014). However, protein complex detection is one of the most challenging problems in PPI network studies, and computational approaches are an appropriate complement for experimental methods to solve this problem. A protein complex contains multiple proteins which are simultaneously linked by non-covalent protein–protein interactions at the same location to perform a functional process in the cell, such as the transcription of DNA, the translation of mRNA, signal transduction and the cell cycle (Li et al., 2010; Moschopoulos et al., 2011).
Generally, PPI networks are modeled as a graph wherein each vertex and edge represents a protein and an interaction between two proteins, respectively (Srihari and Leong, 2013). There are several computational methods to predict protein complexes in PPI networks. The MCL (Enright et al., 2002) method clusters PPI networks by simulating random walks. Some methods such as MCODE (Bader and Hogue, 2003) predict dense sub-graphs as protein complexes. The restricted neighborhood search clustering algorithm (RNSC) (King et al., 2004) is a cost-based algorithm that searches for clusters with minimum cost in the network. The LCMA (Li et al., 2005), CFinder (Adamcsek et al., 2006) and CMC (Liu et al., 2009) algorithms detect protein complexes based on clique merging. The CFA (Habibi et al., 2010) algorithm predicts k-connected sub-graphs. GA-PPI (Pizzuti and Rombo, 2012, 2013) applies a genetc algorithm with different topology-based fitness functions to cluster PPI networks. The integrative hierarchical clustering algorithm (Wu et al., 2013) integrates multiple heterogeneous data such as Gene Ontology (GO) terms, gene expression profiles, tandem affinity purification with mass spectrometry data and PPI data to detect protein complexes in a hierarchical clustering approach.
Based on studies in yeast (Dezső et al., 2003; Gavin et al., 2006), each complex is composed of a core and its attachments. The core is the central functional unit of a complex. Attachment proteins are bound to the core to complete its function (Srihari and Leong, 2013). In the core, proteins have relatively more interactions with each other. Accordingly, such methods as CORE (Leung et al., 2009) and COACH (Wu et al., 2009) predict cores and attachments in two separate steps.
Recently, researchers have tried to improve the accuracy of complex prediction methods by integrating biological information with PPI data. Feng et al. (2011) and Chen and Yuan (2006) applied microarray data, while Ozawa et al. (2010) and Ma et al. (2012) combined domain-domain interaction information with PPI data.
In the last few years, GO annotations have also been applied to improve the accuracy of complex prediction (Mukhopadhyay et al., 2012; Wang et al., 2012b). The GO database is composed of GO terms and their relationships. GO terms are grouped into three domains: biological processes (BP), molecular functions (MF) and cellular components (CC) (Zhang, 2009; Wang et al., 2012b). Each gene or protein is annotated to one or more GO term(s) and each term is related to one or more other terms in a directed acyclic graph (DAG) structure. According to the experimental observation that proteins in a complex tend to carry out common biological functions (Dezső et al., 2003), some methods use GO annotation to measure the reliability of interaction between each pair of proteins. In these methods, low-reliability interactions are first removed to predict complexes in the new unweighted purified network (Wang et al., 2012b). Although PPI networks are purified by GO information, these data are not considered for complex detection in the algorithm. In other words, an unweighted network is given as an input to the algorithm.
In this article, the semantic similarity between protein pairs is calculated by the Lin method (GraSM) to weigh the PPI networks (Couto et al., 2007). We develop the WCOACH (Weighted COACH) method based on the COACH algorithm and predict protein complexes in weighted PPI networks. The proposed algorithm and such well-known algorithms as COACH, CORE, ClusterOne (Nepusz et al., 2012), MCODE, HC-PIN (Wang et al., 2011), OH-PIN (Wang et al., 2012a) and IPCA (Li et al., 2008) are then run on five PPI networks of yeast including DIP (Xenarios et al., 2002), Krogan (Krogan et al., 2006), MIPS (Mewes et al., 2006), Gavin 2002 (Gavin et al., 2002) and Gavin 2006 (Gavin et al., 2006). The predicted complexes are compared to the experimentally detected complexes of CYC2008 (Pu et al., 2009). The results show that WCOACH can predict protein complexes with high performance.
A weighted PPI network is shown as a weighted graph G = (V, E, W) where V, E and W denote the set of vertices (proteins), edges (interactions), and weight function W:E→R mapping an edge to a real number. The weight of each interaction is a criterion to show its reliability. A neighborhood graph of a vertex v is defined as
Although COACH (COre-AttaCHment based method) is a well-known algorithm for protein complex prediction in unweighted PPI networks, it predicts cores and attachments as complexes without considering any difference between interactions with high or low semantic similarities. Therefore, considering weighted graphs in the COACH method can increasingly enhance the accuracy of complex prediction. In this paper a novel algorithm, called WCOACH, is represented to support weighted networks for complex prediction. The main steps of the proposed algorithm are as follows:
1. Weigh a PPI network based on GO.
2. Detect preliminary cores.
3. Remove redundant cores.
4. Add attachment proteins to each core for constructing a predicted complex.
The details of the algorithm are explained in the next sub-sections.
Calculation of semantic similarity between protein pairsThe GO database, created from GO terms, forms a DAG based on their relationships to each other. In this paper, we considered three kinds of relationships between terms, called “is-a”, “part-of” and “regulates”, representing specific-to-general, part-to-whole and regulation relationships, respectively. The semantic similarity between terms is defined based on DAG structure (Zhang, 2009).
Several methods have been developed to measure the semantic similarity between GO terms. Structure-based methods (Wu and Palmer, 1994; Leacock and Chodorow, 1998) measure the semantic similarity based on path length and common parentage. Information content-based methods such as those of Lin (1998) and Resnik (1995) measure the semantic similarity based on a priori probabilities or information content of GO terms (Zhang, 2009). In this article, the csbl.go package (http://csbi.ltdk.helsinki.fi/csbl.go) is employed to measure the semantic similarity between protein pairs based on the Lin (GraSM) method. It is clear that the interaction between each pair of proteins unannotated in GO is not weighed. Therefore, the Acceptance-Rejection method is applied to generate values based on weight distribution of other interactions. These values are considered as the weight of unannotated interactions.
We downloaded the GO annotation file from http://www.yeastgenome.org/download-data/curation with version 2, dated 01/18/2014.
Protein complex predictionBased on the COACH method, a new method called WCOACH is developed to predict protein complexes in weighted PPI networks. A weighted graph G = (V, E, W) is given as an input to the algorithm. The set Φ is defined as follows:

Predict preliminary cores
To make the set of final cores Γ from Π the “Redundancy-filtering” procedure of the COACH method is utilized (see Algorithm 2), wherein τ is a threshold to control the overlap between predicted cores (see section 4.3).

Remove redundant cores
In the COACH method, a protein is considered as an attachment if it interacts with more than half of the proteins in the core. In real complexes (Gavin et al., 2006), many attachment proteins interact with fewer than half of the proteins in the core. The COACH method cannot detect these attachments. For example, as shown in Fig. 1, the core consists of five proteins in the Coatomer COPII complex. There are six attachments in this complex; however, only one of them interacts with more than two proteins of the core. Our goal is to improve the COACH method for detecting these attachments through defining weighted closeness.

Coatomer COPII complex. Proteins shown in the gray circle form the core and other proteins are attachments. The COACH method cannot detect YJL034W, YGR116W, YDR517W, YBR192W or YGR155W proteins as attachments because they interact with fewer than half of the core proteins.
The attachments set of core C = (VC, EC, WC) is defined as:
In this section, we introduce the PPI networks applied to test our method. Some evaluation criteria are then represented to compare the WCOACH method to the other methods. After that, we show how to compute the neighborhood affinity threshold (τ). Finally, we compare our method to the others on the PPI networks based on defined evaluation criteria.
DatasetsIn this article, five PPI networks of yeast are applied to compare our method with others: DIP (Xenarios et al., 2002), Krogan (Krogan et al., 2006), MIPS (Mewes et al., 2006), Gavin 2002 (Gavin et al., 2002) and Gavin 2006 (Gavin et al., 2006). The properties of these networks are shown in Table 1. The real complexes dataset CYC2008, with 428 protein complexes, is used as a benchmark for evaluation (Pu et al., 2009).
| Network name | Number of proteins | Number of interactions |
|---|---|---|
| DIP | 4930 | 17201 |
| Krogan | 2675 | 7084 |
| Gavin 2002 | 1352 | 3210 |
| Gavin 2006 | 1430 | 6531 |
| MIPS | 4564 | 15175 |
To compare WCOACH with the other methods, precision, recall, F-measure, coverage rate and P-value are introduced as evaluation criteria.
Let ℘ = {P1, P2, …, Pk} and ℜ = {R1, R2, …Rm} be two sets of predicted and real complexes, respectively. Precision and recall are represented as follows (Ahn et al., 2013; Zaki et al., 2013):
For the predicted and real complex sets ℘ = {P1, P2, …, Pk} and ℜ = {R1, R2, …Rm}, coverage rate is defined as follows (Ma and Gao, 2012):
The biological significance of the predicted complexes is evaluated by P-value. Given a predicted complex P = (VP, EP), that includes k proteins from a functional group (such as a GO term) with size m, the P-value of P is defined as (Ma and Gao, 2012):
The P-value is the probability that a given set of proteins is enriched by a given functional group merely by chance (Ma and Gao, 2012). Accordingly, a low P-value shows high statistical significance. Here, the GO Term Finder tool (Boyle et al., 2004), available on http://go.princeton.edu/cgi-bin/GOTermFinder, is used to calculate P-values of predicted protein complexes.
Neighborhood affinity thresholdThe neighborhood affinity threshold (τ ∈[0,1]), used in Algorithm 2, controls the overlap between predicted cores. A zero (one) value of τ shows there is no overlap (there is overlap) between cores. To find the appropriate threshold, WCOACH is run with various values of τ on the Gavin 2006 network. Fig. 2 shows that an increase in τ causes higher F-measure and coverage rate values. According to this experiment, τ is set to 0.85 as default.

The F-measure and coverage rate values of WCOACH for various values of neighborhood affinity threshold (τ) on the Gavin 2006 network.
In this section, the method is compared to the COACH (Wu et al., 2009), ClusterOne (Nepusz et al., 2012), CORE (Leung et al., 2009), MCODE (Bader and Hogue, 2003), HC-PIN (Wang et al., 2011), OH-PIN (Wang et al., 2012a) and IPCA (Li et al., 2008) algorithms. In this experiment, all complexes with fewer than three proteins are ignored.
All algorithms were run on the DIP, Gavin 2002, MIPS and Krogan networks. Their precision, recall and F-measure values are shown in Fig. 3. It can be seen that WCOACH has a high performance on the DIP, Krogan and Gavin 2002 networks and its precision, recall and F-measure values are higher than those of the other algorithms. Note that in our experiment, the HC-PIN algorithm detected only eight clusters on the DIP network and its precision, recall and F-measure values are very low; for this reason, this algorithm is ignored on the DIP network. On the MIPS network, WCOACH displays the highest F-measure value after CORE and the recall value of our algorithm is higher than COACH, CORE, OH-PIN, HC-PIN and MCODE.

Precision, recall and F-measure values of various algorithms on the Krogan, DIP, Gavin 2002 and MIPS networks.
The coverage rates of the algorithms are shown in Fig. 4. WCOACH displays the highest coverage rate on the DIP network. Our method also has acceptable coverage rates on three other networks.
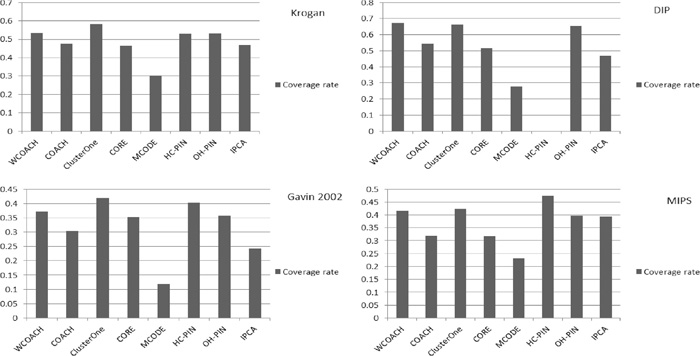
Coverage rate values of various algorithms on the Krogan, DIP, Gavin 2002 and MIPS networks.
To evaluate the biological significance of this method, the P-values of all predicted clusters on the DIP network were calculated. The GO Term Finder tool was applied to calculate P-values based on the BP and MF domains of GO. In this experiment, a predicted cluster P is significant if P – value (P) < ρ where ρ is a cut-off for the P-value. The percentage of significant clusters for several values of ρ is shown in Figs. 5 and 6, where the P-values are calculated based on BP and MF, respectively. With respect to using GO for protein complex prediction, the WCOACH method detected more clusters with low P-values.

Percentage of significant clusters for various values of P-value cut-off calculated based on the BP domain.

Percentage of significant clusters for various values of P-value cut-off calculated based on the MF domain.
In this paper, the COACH method is modified and a new weighted core attachment method, WCOACH, is proposed to detect protein complexes in weighted PPI networks. The semantic similarity measure between protein pairs based on GO structure is applied to estimate the reliability of protein interactions. This measure, calculated by the Lin (GraSM) method, is considered as the weight of each interaction. The COACH method cannot detect all attachment proteins in complexes. In the WCOACH method, the weight of interactions is applied to detect attachment proteins.
We would like to thank Dr. C. N. Moschopoulos for providing PPI datasets.