Chemical and Physical Analysis
Change of State of Lime Phase and Inhibition of Hydration Reaction by Coexisting Oxides in Steelmaking Slag
2021 年 61 巻 5 号 p. 1594-1602
詳細
2021 年 61 巻 5 号 p. 1594-1602
To promote the utilization of steelmaking slag, the hydration reactivity and electronic state of free lime (CaO) were observed in this study. It was confirmed by X-ray diffraction (XRD) and thermal analysis that the hydration reactivity decreases when CaO forms a lime-phase solid solution with FeO, and it was revealed by X-ray photoelectron spectroscopy (XPS) observations that the decrease in reactivity occurs due to deepening of the energy levels of the electron orbitals. The morphology and hydration reactivity of the free lime contained in a synthesized slag were observed by XRD and electron probe microanalysis. Fe-containing phases and double oxides were found to be adjacent to free lime; the former is considered to be related to the formation of a solid solution with FeO, and the latter closely surrounds the free lime and shields it from the outside; thus, both contribute to suppressing the hydration reaction. Furthermore, it was found that simply mixing lime with double oxides suppressed the hydration reaction. XPS and thermal analysis confirmed that a chemical change occurred on the surface of the free lime; that is, the energy levels of the electron orbitals are deepened and the hydration reaction is suppressed due to chemical interactions with the double oxides to stabilize the lime surface. It was clarified that in order to effectively promote the hydration of free lime, it must be exposed by breaking the structure of the slag that protects the free lime and suppresses hydration.
A large amount of iron and steel slags, approximately 40 million tons, as byproducts of the steel manufacturing process is generated per year in Japan and used as recycled materials for construction and civil engineering. Steelmaking slag, which accounts for 14 million tons, is generated from the production of steel from pig iron, and most of it is used in land areas such as civil engineering and roadbeds.1) Due to its chemical nature, it has recently been proposed to use converter slag, a kind of steelmaking slag produced by converters, to restore paddy fields damaged by the Tsunami of the Great East Japan Earthquake.2) In addition to the physical properties of these materials such as strength and chemical properties such as pH, the hydration expansion behavior is also important. It is known that lime added to separate impurities in the steelmaking process can become free lime (CaO), and hydration expansion (CaO → Ca(OH)2) may occur.3,4) To evaluate the hydration and swelling properties of slag and perform appropriate aging treatments, methods for quantifying the free lime contained in slag are being investigated. A method recommended by the Iron and Steel Institute of Japan has been established as a quantitative process by extraction with ethylene glycol.5) A method combined with thermogravimetric analysis has also been proposed to enable differential quantification with Ca(OH)2, which is a cause of inaccuracy in the ethylene glycol quantification method.6) Since CaO reacts with ethylene glycol to thicken and gel, it has been reported that the addition of halogenated alcohols can suppress the changes in its physical properties.7) Quantitative determinations by X-ray diffraction (XRD) have also been reported,8,9,10,11) and there has been a study showing that the free lime content can be determined by calorimetric measurements.12,13) It has also been reported that the intensities of cathodoluminescence and X-ray excited photoluminescence are correlated with the amount of free lime in slag.14)
Although it is possible to determine the free lime content, the expansion characteristics and related mechanisms of slag have not been sufficiently clarified, and there is a poor correlation between the quantitative free lime content and expansion characteristics. It is known that the hydration of calcium silicate, calcium aluminate, and magnesium oxide15,16,17) is not negligible. Furthermore, divalent metal oxides such as FeO and MnO can form solid solutions in CaO, and the hydration reactivity of free lime changes depending on the solid solubility (the amount of divalent metal oxide in the solid solution).3,4,13) Such solid solutions have been confirmed by electron probe microanalysis (EPMA) element distribution images, and it has been shown that they can be identified by the cathodoluminescence method.18)
Previously, we synthesized a lime-phase solid solution and investigated its hydration reactivity; we also carried out X-ray diffraction (XRD) measurements to estimate the amount of free lime contained in the slag, as well as the solid solubility of the divalent metal oxides in the lime phase contained in the slag.10,11) However, the hydration behavior and expandability of the free lime contained in the slag were considered to be different from that of the lime phase alone, and so in this study, simulated slag was synthesized and analyzed. The solid solution formation of free lime in the slag was verified, and the type and distribution of the crystalline phases co-included with the free lime were observed by powder XRD and EPMA. The chemical effects of the oxide phases (calcium silicate and calcium ferrite) surrounding the free lime were also investigated by X-ray photoelectron spectroscopy (XPS).
Solid solutions of CaO and FeO, namely (Ca, Fe)O, were synthesized by a high-temperature solid-state reaction. Here, the compositional formula is denoted as Ca1−xFexO (0 < x ≤ 0.10). As a pretreatment, CaO powder (Fujifilm Wako Pure Chemicals, 99.9%) was calcined at 1123 K for more than 2 h in air to remove Ca(OH)2. Specific ratios of FeO powder (Kojundo Chemical Lab., 99.5%) and CaO were homogeneously mixed depending on the desired composition (x), and the resulting mixture was pelletized. The pellet was fired in an infrared lamp heating furnace (ULVAC, MILA-3000) at 1373 K for 7 h under an inert gas atmosphere by filling with 1 atm of pure N2 gas (>99.9995%) to prevent oxidation of the sample. A list of all the samples synthesized in this paper is provided in Table 1 together with the synthesis conditions and hydration procedure.
| Sample | Sample type | Raw materials | Heat treatment | Analysis | Hydration test | |
|---|---|---|---|---|---|---|
| A | CaO (x = 0) | – | XPS | Steam aging of powder sample at 363 K→XRD, TGA | ||
| B | Solid solutions Ca1−xFexO | x=0.03 | CaO and FeO | 1373 K for 7 h 1 atm N2 | – | |
| C | x=0.06 | |||||
| D | x=0.10 | XPS | – | |||
| E | Simulated slag | CaO, FeO, SiO2 and MgO (mass ratio 50:22:16:12), | 1823 K for 0.5 h and 1773 K for 2 h argon flow | XRD, EPMA | Steam aging of block sample at 363 K for 24 h→XRD | |
| F | CaO | – | – | Stored in a desiccator as pellet →XRD | ||
| G | composite | CaO and Ca2Fe2O5 (mass ratio 1:1) | ||||
| H | 1273 K for 4 h | XPS, DSC | – | |||
| I | CaO and Ca2SiO4 (mass ratio 1:1) | |||||
XRD patterns of the processed materials were obtained using a powder X-ray diffractometer (Rigaku, MiniFlex600, Cu Kα, 40 kV, 15 mA, graphite crystal monochromator) to verify the formation of the solid solutions. The lattice parameters a were determined from the diffraction lines. The solid solubilities, as indicated by the mole fraction x, were determined from a according to Eq. (1).
| (1) 9) |
The electronic states of the solid solutions were analyzed by XPS (Surface Science Instruments, SSX-100, Al Kα, 10 kV). Pellets after firing were used as the sample, and the surface was not etched by sputtering ions. As the samples were insulators, a neutralizing gun was used to remove the effects of charging. The energies of each spectrum were calibrated by the energy of the C 1s spectrum from the impurity carbon attached to the surface of each sample.
2.1.2. Reactivity of the Lime-phase Solid SolutionsEach solid solution sample (sample A–D in Table 1) was crushed into a powder, and the hydration reactivity was compared by exposure to water vapor. Water was boiled in a flask connected to a reflux condenser, and the powders were placed in a crucible in the steam at 363 K. The steam-aging treatment was performed with stirring of the sample in the crucible at intervals of 30 min. The XRD patterns were measured for samples taken at regular intervals. The hydration rates were determined by simultaneous thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements (METTLER TOLEDO, TGA/DSC1, 200 mL/min argon flow) based on weight loss due to heat dehydration.
2.2. Synthesis of the Simulated SlagPowders of CaO, FeO (Kojundo Chemical Lab., 99.5%), SiO2 (Fujifilm Wako Pure Chemical, 99.9%), and MgO (Fujifilm Wako Pure Chemicals, 99.9%) were mixed in a mortar at a mass ratio of 50:22:16:12 and pelletized using a press. The pellets were placed in an MgO crucible and heat-treated under an argon flow (200 mL/min) in a high-frequency induction furnace using carbon as a heat-generating dielectric. The temperature was maintained at 1823 K for 30 min, then decreased to 1773 K and heated for 2 h. After that, the sample was gradually cooled at a rate of −100 K/h. This sample was labeled sample E.
The obtained block slag was surface-polished, then gold-coated for conductivity and observed by EPMA (JEOL, JXA-8200, accelerating voltage 15 kV, electron beam diameter 1 μm). XRD measurements were performed using powder to analyze the crystal phases and calculate the solid solubility of the free-lime-phase solid solution. The block slag was exposed to water vapor with the same setup as that described in section 2.1.2, taken out 24 h later, and subjected to XRD measurements to determine the progress of the hydration reaction.
2.3. Evaluation of the Effects of Adjacent Double Oxides 2.3.1. Synthesis of the Double OxidesCaO and α-Fe2O3 (Rare Metallic, 99.99%) were mixed at a molar ratio of 2:1, pressed into pellets with a diameter of 10Φ, and heated at 1273 K for more than 12 h in a high-temperature electric furnace to synthesize Ca2Fe2O5. The same treatment was carried out for a powder mixture of CaO and SiO2 at a molar ratio of 2:1 to synthesize β-Ca2SiO4.
2.3.2. Analysis of the Composite Samples of Double Oxides and CaOThe following three types of composite samples were tested.
(a) CaO and the Ca2Fe2O5 powder, synthesized as described in section 2.3.1, were mixed at a mass ratio of 1:1, and a pellet with a diameter of 10Φ was formed at 400 MPa using a high-pressure press. This pellet (sample G) was stored in a desiccator together with a pellet made of pure CaO pressed under the same conditions (sample F), and after a certain period of time, XRD measurements were performed to compare the progress of hydration.
(b) Pellets prepared and molded in the same manner as described in (a) were heated in a high-temperature electric furnace for 24 h at 1273 K (sample H). After confirming that no changes occurred in the crystal phases by XRD, XPS measurements (Al Kα, 6 kV) of the O 1s and Ca 2p orbitals were performed in pellet form. The surface was not etched by sputtering ions. A neutralizing gun was used to remove the effects of charging. The energies of each spectrum were calibrated by the energy of the C 1s spectrum from the impurity carbon attached to the surface of each sample. The XPS data of CaO, Ca(OH)2, β-Ca2SiO4, and Ca2Fe2O5 as reference samples were also measured. Subsequently, the dehydration temperature of the Ca(OH)2 generated by reacting naturally with moisture in the air was measured by DSC.
(c) CaO and the β-Ca2SiO4 powder, synthesized as described in section 2.3.1, were mixed at a mass ratio of 1:1, and a press machine was used to form pellets with a diameter of 10Φ at 400 MPa, which were heated in a high-temperature electric furnace at 1273 K for 24 h (sample I). XRD, XPS, and thermal analysis were performed as described in (b).
The XRD diffraction lines of the obtained Ca1−xFexO were shifted compared with those of pure CaO based on the composition of each sample, that is, the value of x. In other words, the formation of solid solutions was confirmed, and from the lattice constants, the x values were calculated.
The XPS results of the solid solution (sample D) and pure CaO (sample A) are shown in Fig. 1 for comparison. The binding energies of Ca 2s, 2p, 3s, 3p, and O 1s tended to shift to higher energies with the addition of FeO. This phenomenon likely occurred because Fe has a higher electronegativity than Ca and tends to attract electrons more strongly, and thus FeO has a stronger covalent bond than CaO. Therefore, the addition of Fe reduces the electron density around Ca and deepens the energy level of the Ca electron orbital. A similar situation will occur for O 1s. Shifts with the same tendency have been confirmed with good reproducibility in other solid solutions.

XPS peaks of CaO and Ca0.9Fe0.1O: (a) Ca 2s, (b) Ca 2p, (c) Ca 3s and 3p, and (d) O 1s. (Online version in color.)
The molecular orbitals were calculated using the DV-Xα method, and the density of states of each orbital was compared, as shown in Fig. 2. Here, CaO, FeO, and their solid solutions were calculated using the Ca13O14, Fe13O14, and Ca12FeO14 cluster models, respectively. Each cluster, which has a metal ion as the center and includes up to the third adjacent ions of the rock-salt-type structure, was calculated by considering the Madelung potential. In the solid solution model, one of the second-closest metal ions was exchanged from Ca2+ to Fe2+. In FeO, the 3d, 4s, and 4p orbitals of Fe overlapped with the occupied electron bands consisting mainly of the 2s and 2p orbitals of O, confirming the covalent character. In CaO, orbital overlap between O 2s, 2p, and Ca was observed, but the ratio was small and the covalent bond was weak. In FeO and the solid solutions, the band mainly composed of the Fe 3d orbital formed the upper end (Fermi level) of the valence band, and the energy level of the occupied electron band including Ca 3p was deeper than that in CaO. Thus, the calculated molecular orbitals were in good agreement with the XPS observations.

Electronic density of states of (a) CaO, (b) FeO, and (c) their solid solution calculated by DV-Xα. (Online version in color.)
Figure 3 shows the XRD patterns of the powder samples of CaO and the synthesized solid solutions of Ca1−xFexO after the steam-aging treatment. Pure CaO (sample A) was completely hydrated within 15 min and changed to Ca(OH)2, but the solid solutions proceeded to hydrate over a longer period of time. The diffraction line intensities from the solid solutions gradually decreased with aging time, while the Ca(OH)2 diffraction intensities increased as the hydration reaction proceeded. When x = 0.03 (sample B), the diffraction lines of the solid solution disappeared completely after 6 h, indicating completion of the hydration reaction. When x = 0.06 (sample C), the hydration reaction was complete in 26 h. Therefore, the progress of the hydration reaction was significantly delayed due to the increase in solid solubility.
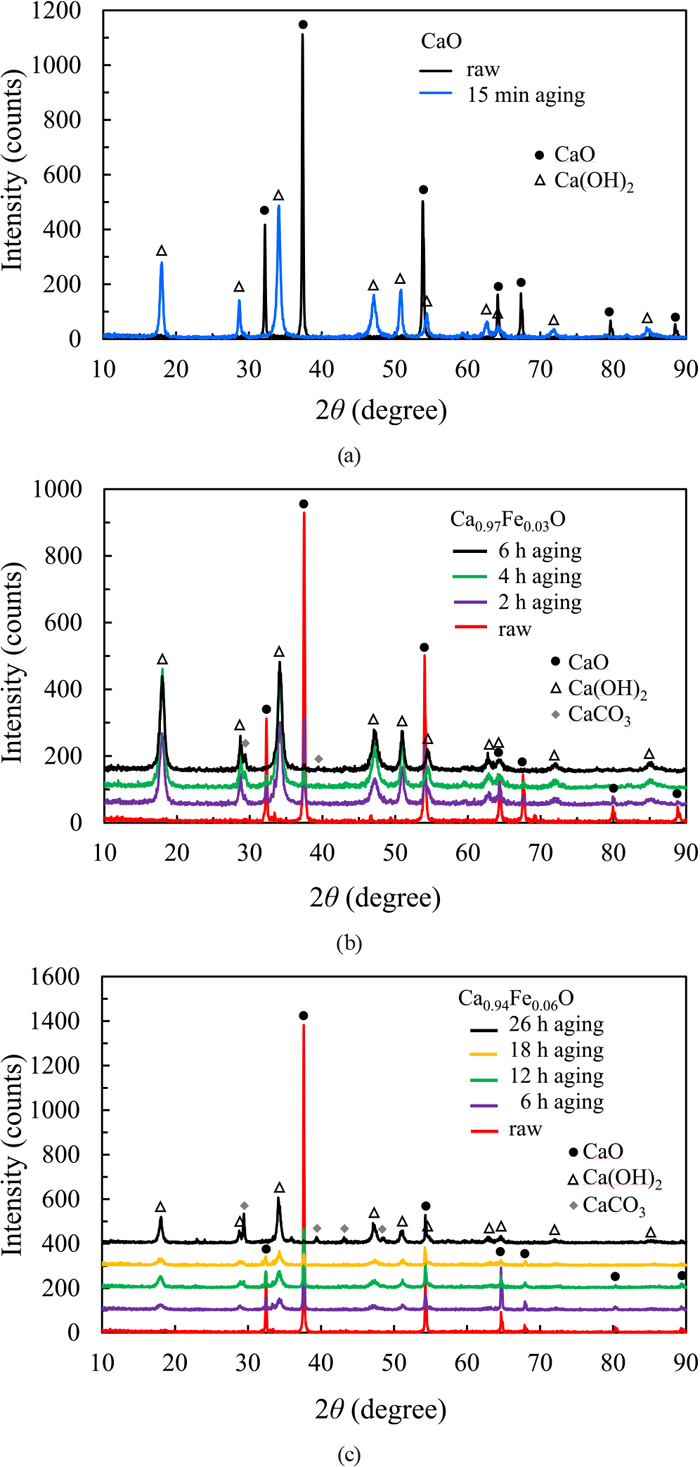
XRD patterns of CaO and the Ca1−xFexO solid solution with steam aging time. (a) CaO, (b) x = 0.03, (c) x = 0.06. (Online version in color.)
The thermal analysis of the hydrated sample showed an endotherm and a decrease in mass due to the dehydration of Ca(OH)2 at approximately 630 to 710 K. Table 2 shows the hydration rates calculated from the dehydration amounts determined by TGA, and the results were compared for each aging time. When x = 0.03 (sample B), approximately half of the sample was hydrated in 90 min; if the reaction were to proceed at this rate, the sample should be completely hydrated in 3 h according to calculations, but it actually required 6 h to complete. The reaction proceeded rapidly at the start of the aging treatment, then slowed with time. The hydration reaction occurs from the surface of the sample, meaning that hydration inside the sample is less likely to occur after a hydration layer forms on the surface. On the other hand, when x = 0.06 (sample C), only 20% of the sample was hydrated after 90 min, confirming that the hydration reaction was suppressed by an increase in the solid solubility. The XPS measurements showed that the addition of Fe reduced the electron density around Ca and deepened the energy levels of the Ca and O electron orbitals. Therefore, it is considered that an increase in Fe content reduces the chemical reactivity and suppresses the hydration reactivity of the system.
| Sample | Solid solubility, x | Aging treatment time (min) | 30 | 60 | 90 |
|---|---|---|---|---|---|
| B | 0.03 | Dehydration amount (mg) | 0.08 | 0.10 | 0.14 |
| Hydration rate (%) | 27 | 35 | 49 | ||
| C | 0.06 | Dehydration amount (mg) | 0.024 | 0.041 | 0.060 |
| Hydration rate (%) | 6.4 | 14 | 20 |
Figure 4 shows the XRD pattern of the slag obtained from the pseudo-quaternary system (sample E). In addition to free lime, larnite (β-Ca2SiO4), α-Fe, and magnesiowüstite ((Mg, Fe)O), in which FeO is dissolved in MgO (free magnesia), were observed. Here, metallic iron was generated because carbon was used as the heating element and the inside of the furnace had a reducing atmosphere. The diffraction lines of the lime phase shifted to higher angles compared with those of pure CaO, clearly indicating that a solid solution formed with FeO. The lattice constant calculated from the 2θ angles of the four diffraction lines with indices 200, 220, 311, and 222, which are not affected by overlap with the diffraction lines from other crystal phases, was a = 0.4797(1) nm, and the solid solubility was calculated as x = 0.045(3) using Eq. (1). Here, CaO can form a solid solution with MgO, but because of the large difference in lattice constants (i.e., the difference between the ionic radii of Ca2+ and Mg2+), such a solid solution does not form easily, and thus CaO was regarded as forming a solid solution with only FeO.

XRD pattern of the slag synthesized from the mixture of CaO, FeO, SiO2, and MgO. (Online version in color.)
Figure 5 shows the observed EPMA images, from which the distribution of each phase identified by XRD was determined based on the contrasting elemental distributions. The distribution of Fe overlapped with the shape of the free lime particles, confirming the formation of a solid solution. The distribution of high-concentration Fe overlapped with that of MgO. It is known that MgO and FeO, which have similar lattice constants that are smaller than that of CaO, easily form a complete solid solution. The angles of the diffraction lines were located near the halfway point between those of MgO and FeO, and they were mixed in a nearly equal ratio. Similar to CaO, MgO is known to cause hydration expansion as free magnesia15,16,17) and has a lower hydration activity than CaO, and hydration proceeds slowly because of the chemical properties of MgO. In addition to the above, it can be understood that the facile formation of a solid solution containing a high concentration of FeO also effectively suppresses the hydration activity.

EPMA images of the pseudo-quaternary slag. (a) Distributions of Ca, Fe, Mg, Si, and O. (b) Distributions of Ca, Fe, and O shown on an enlarged scale and an intensity distribution map along the indicated line. (Online version in color.)
Free lime particles containing FeO with particle sizes up to approximately 30 μm were observed by EPMA. The distribution of Fe within the particles appeared to be relatively uniform, with particles scattered within a matrix of Ca2SiO4 (C2S). Figure 5(b) shows an enlarged view of a free lime particle, illustrating that they can naturally feed FeO from adjacent magnesiowüstite. However, the Fe component cannot dissolve completely because the solid solution limit is small, and the remaining Fe precipitates so as to surround the outer periphery of the free lime particles. The substance is an oxide such as Ca2Fe2O5 (C2F). The absolute amount was too small to be clearly detected by XRD, but it is a commonly found product in slag. According to the pseudo-binary phase diagram of CaO–FeO,19,20) the solid solubility limit of FeO in the lime phase is 12 mol% at 1373 K, and as the proportion of FeO increases, the two phases (solid solution and C2F) coexist. In addition, the melting point of C2F is 1450°C,21) and during the heat treatment at higher temperatures, it becomes a melt and enters the gaps to surround the free lime. The free lime in the slag is surrounded by stable oxides such as C2F and C2S that coexist, and a dense structure is created. As a result, the surface of the free lime in the slag is difficult to expose, and the hydration reaction is suppressed.
3.2.2. Water Vapor Reactivity of the Synthetic SlagWhen XRD measurements were performed after vacuum-drying the slag sample exposed to water vapor (sample E), all of the diffraction line intensities of the lime phase decreased, and Ca(OH)2 diffraction lines appeared, confirming that the hydration reaction proceeded. However, much of the lime phases remained despite 24 h of exposure, as shown in Fig. 6. As shown in Fig. 3(c), the hydration of the solid solution with x = 0.06 (sample C) reached completion in 26 h, whereas the hydration of the lime phase in the slag was incomplete despite the lower solid solubility of the lime phase in the slag (x = 0.045(3), as described above), which seems inconsistent. Based on this result, it is understood that the hydration reaction of free lime in the slag was considerably suppressed.

XRD pattern of synthetic slag after 24 h of exposure to 90°C steam. (Online version in color.)
Figure 7 shows the changes in the XRD patterns of pellets of CaO (sample F) and CaO mixed with Ca2Fe2O5 (sample G) up to 72 h after hydration began. Hydration of the CaO pellet gradually progressed, and a distinct diffraction line corresponding to Ca(OH)2 was detected after 72 h. On the other hand, in the mixed pellet, the baseline intensity was slightly higher, but the Ca(OH)2 diffraction line could not be clearly observed. Although a slight diffraction line was confirmed, its intensity was clearly weak, indicating a difference in hydration reactivity. The DSC measurement, as described in section 3.3.2, suggests that a chemical reaction occurs at the interface between the double oxide and CaO. This DSC change can be confirmed by measuring a sample in which CaO and the double oxide are simply mixed. It is considered that the chemical reaction at the interface proceeds even at room temperature due to the high reactivity of CaO.

Observation of hydration reaction of CaO pellets by XRD. (a) CaO and (b) CaO/Ca2Fe2O5 mixed sample. (Online version in color.)
The pellet prepared by heating the mixture of CaO and Ca2Fe2O5 in a high-temperature electric furnace for 24 h at 1273 K (sample H) was analyzed by XRD. In this composition, the phase diagram22,23) shows that the two phases coexist, and the XRD pattern shows a simple mixture of both phases without the formation of a third. The same was observed for a mixed sample of CaO and β–Ca2SiO4 (sample I), which was consistent with the reported phase diagram.22)
The XPS O 1s and Ca 2p results of CaO/Ca2Fe2O5 (sample H) shown in Fig. 8 illustrate that the binding energies of the mixed sample shifted to slightly higher energies compared with the reference spectra of the individual components. In other words, the spectra are not simply superpositions of the spectra of CaO and Ca2Fe2O5, but instead correspond to another state. Similarly, Fig. 9 shows the XPS of the mixture with β–Ca2SiO4 (sample I). As confirmed by XRD, if there is no chemical reaction between CaO and the double oxide, the spectrum of the mixed sample should be the sum of the spectra of the individual components. However, the spectral shapes appear to be different. Therefore, it is considered that reactions occurred at the contact surface between CaO and the double oxide and were detected by XPS, which is surface sensitive.

XPS of the CaO/Ca2Fe2O5 mixed sample compared with the reference spectra of CaO and Ca2Fe2O5. (a) Ca 2p and (b) O 1s. (Online version in color.)

XPS of CaO/β–Ca2SiO4 mixed sample compared with the reference spectra of CaO and β–Ca2SiO4. (a) Ca 2p and (b) O 1s. (Online version in color.)
Figure 10 shows the DSC results of both mixed samples, which demonstrate the dehydration temperature of the contained Ca(OH)2. Here, the mixed samples were compared with CaO, Ca(OH)2, and the double oxide. For the CaO and CaO/double oxide mixed samples, the dehydration of Ca(OH)2 caused by hydration in air before the measurements was performed. However, even with the same substance, Ca(OH)2, there was a difference in the endothermic peak temperature. It is known that Ca(OH)2 produced industrially as a reagent and Ca(OH)2 produced from CaO by absorbing water from air have different binding forces with H2O.24) In terms of DSC, Ca(OH)2 as an industrial product has a stronger binding force, and therefore the endothermic peak is on the high-temperature side. The endothermic peak temperature of the CaO/double oxide sample was considered to be equal to that of CaO but shifted to higher temperatures, indicating a difference in the binding force between Ca2+ and H2O. This is expected to be attributable to the chemical action between CaO and the double oxide, and was assumed to change the state of the produced Ca(OH)2 on the CaO surface. It should be noted that such a temperature difference was observed even in a powder sample in which CaO and the double oxide were simply mixed, and thus it is considered that the interaction occurs on the surface without heat treatment.

Differential thermal calorimetry of CaO/double oxide mixed samples compared with the reference spectra of CaO, Ca(OH)2, and the corresponding double oxides. (a) CaO/Ca2Fe2O5 and (b) CaO/β–Ca2SiO4. (Online version in color.)
As described above, the higher-energy shift of the XPS peaks of CaO mixed with the double oxides indicates that the number of electrons tends to decrease. CaO has a rock-salt-type crystal structure where Ca2+ is hexacoordinated to O2−, but the ionic radius of Ca2+ is large and this crystal structure is unstable, making CaO highly reactive.25) Therefore, CaO and the double oxides react to increase the coordination number of Ca2+, and as a result, the interactions with surrounding atoms increase and the electron density around Ca2+ tends to decrease. In addition, considering that divalent Ca on the surface of CaO is replaced by trivalent Fe or tetravalent Si from double oxides, the number of electrons also tends to decrease. Furthermore, DV-Xα calculation results for the Ca12SiO14 cluster in which Ca2+ was replaced by Si4+ also showed an increase in binding energy. Such a deep energy level leads to a decrease in chemical reactivity. Therefore, the lime phase in the slag is not only physically shielded from water by the surrounding dense double oxides to prevent hydration, but the hydration reactivity is also suppressed by a chemical change on the surface.
To evaluate the hydration reactivity of free lime contained in steelmaking slag, a solid solution with FeO (Ca1−xFexO, 0 < x ≤ 0.10) was synthesized by a high-temperature solid-state reaction, its electronic states were analyzed by XPS, and the progress of the hydration reaction was observed by XRD and TGA/DSC. It was confirmed that the hydration reaction slowed as the solid solubility increased, and it was argued that the reason was that the reactivity decreased as the energy state of the electron orbitals became deeper. Slag with a simple composition was synthesized, and the morphology and hydration reactivity of the contained free lime were observed by XRD and EPMA analyses. The free lime in the slag is chemically suppressed by forming a lime-phase solid solution of Ca1−xFexO. At the same time, it is physically shielded from water by the surrounding stable double oxide phases, which is thought to hinder the progress of the hydration reaction. In addition, it was concluded that the coexistence of the lime phase with double oxide phases stabilizes the lime surface by a chemical effect and suppresses the hydration reaction. Therefore, in order to understand the hydration reactivity and expansion behavior of free lime, it is necessary to identify the form and state of free lime present in the slag. Further, in order to efficiently hydrate the free lime by aging treatment, it is desirable to expose the surface of the free lime and bring it into contact with water.