Abstract
Edible burdock (Arctium lappa L.) accumulates an inulin-type fructan. Inulin-type fructan in plant has been hydrolyzed by fructan 1-exohydrolases (1-FEH). We have previously reported on the cloning of aleh1, which encodes a 1-FEH in edible burdock. Here, we describe the cloning of aleh2, which encodes a 1-FEH isozyme in edible burdock, and the functional analysis of the recombinant protein of aleh2 (rAlEH2) that displays properties different from the aleh1 recombinant protein. A cDNA, named aleh2, was obtained by the RACE. The rAlEH2, which was produced by Pichia pastoris, showed 1-FEH activity. Unlike the recombinant protein of aleh1, the rAlEH2 is a 1-FEH enzyme that efficiently hydrolyzes longer-chain fructans than 1-kestose, such as nystose, fructosylnystose and inulin. The expression study in burdock revealed the induction of aleh1 and aleh2 genes by low temperature. These findings indicated that two 1-FEH isoforms were involved in the degradation of the fructan in burdock roots during low-temperature storage.
Abbreviations
IOS, inulooligosaccharides; FOS, fructooligosaccharides; 1-FEH, fructan 1-exohydrolase; 6-FEH, fructan 6-exohydrolase; 1-FFT, fructan:fructan 1-fructosyltransferase; 1-SST, sucrose:sucrose 1-fructosyltransferase; DP, degree of polymerization; HPAEC, high performance anion-exchange chromatography; PCR, polymerase chain reaction; RACE, rapid amplification of cDNA ends.
INTRODUCTION
Edible burdock (Arctium lappa L.), a biennial plant from the family Asteraceae, is regarded by the Japanese as a nutritious food rich in dietary fiber. The roots of edible burdock accumulate inulin-type fructan as a storage carbohydrate. The inulin-type fructan [1F(1-β-D-fructofuranosyl)m sucrose], a β-2, 1-linked fructofuranosyl unit terminated by glucose, accumulates mainly in Asteraceae plants. The inulin-type fructan is synthesized from sucrose by sucrose:sucrose 1-fructosyltransferase (1-SST, EC 2.4.1.99) and fructan:fructan 1-fructosyltransferase (1-FFT, EC 2.4.1.100).1) 2) 3) 4) 5) 6) 7) We have previously reported that 1-FFT is involved in the synthesis of the inulin-type fructan from edible burdock,8) and that inulooligosaccharides (IOS), as well as the inulin-type fructan, were detected in burdock roots stored in soil for six months during winter.9) The amounts of the IOS and fructooligosaccharides (FOS), which are fructans with a low degree of polymerization, increased as the inulin amounts decreased during low-temperature storage after harvest.10) 11) The increase in these oligosaccharides was attributed to be the hydrolysis of the fructans and the action of 1-FFT.8) 9) The production of IOS and FOS in plants during post-harvest and growing seasons has been reported for other plants, including chicory and Jerusalem artichoke.12) 13)
Plant fructans are degraded by fructan exohydrolases (FEHs). Three different types of FEHs, fructan 1-exohydrolase (1-FEH, EC 3.2.1.153),14) 15) 16) 17) 18) fructan 6-exohydrolase (6-FEH, EC 3.2.1.154),19) and 6&1-FEH20) have been reported. These FEHs are distinguished from invertase (β-fructofuranosidase, EC 3.2.1.26) by their inability to hydrolyze sucrose.21) The inulin-type fructan is hydrolyzed by 1-FEH, which preferentially hydrolyzes the β-2,1-linkage of the terminal fructosyl residue of fructans.14) 15) 16) 17) 18) Three 1-FEH cDNAs (1-FEH I, 1-FEH IIa/IIb) have been identified in chicory.14) 15) FEHs belong to family 32 of the glycoside hydrolases (GH32).22) The three-dimensional structures of 1-FEH IIa from chicory has been demonstrated by X-ray crystallography.23) In other plants, such as wheat or perennial ryegrass, several genes encoding FEHs have also been identified.16) 17) 18) 19) 20) Chicory 1-FEH I and 1-FEH II are highly expressed in cold stored roots, and rapid induction of 1-FEH II is caused by defoliation.14) 15) 21) 24) Low-temperature induces gene expression of FEH are known in other plants.25) 26) 27)
Recently, we reported on the cloning of aleh1, which encodes a 1-FEH involved in the degradation of an inulin-type fructan in edible burdock, and functional analysis of the recombinant protein of aleh1.17) However, the genes of other FEHs in edible burdock are yet to be realized. Here, we describe the cloning of aleh2, which encodes a 1-FEH in edible burdock, and functional analysis of the recombinant protein of aleh2 that displays properties different from the recombinant protein of aleh1. Our results suggest that fructans in burdock stored at 0°C are degraded by the combined action of two 1-FEHs induced by low temperatures.
MATERIALS AND METHODS
Plant material. Edible burdock roots (Arctium lappa L.) were freshly harvested in November from an experimental field of the Makubetsu Agricultural Co-operative, Hokkaido, Japan and then stored in soil at 1 m depth until May. After washing with water, the roots were stored at -80°C until further use. For gene expression analysis, samples were used in the same roots described in a previous study.10) 11) These roots were also stored at -80°C until further use.
Substrates. Saccharides used as substrates were prepared as follows: Crystalline 1-kestose [1F-β-D-fructofuranosylsucrose, 1-kestotriose] and nystose [1F(1-β-D-fructofuranosyl)2 sucrose, 1, 1-kestotetraose] were prepared from sucrose using Scopulariopsis brevicaulis β-fructofuranosidase.28) Fructosylnystose was prepared from Jerusalem artichoke tubers in our laboratory. Inulobiose and inulotriose were prepared from burdock roots that were stored for six months.9) Sucrose and inulin (MW 5,000) were purchased from Wako Pure Chemicals Industries (Osaka, Japan). Neokestose was prepared from asparagus roots as described previously.29) 6-Kestose was provided by Dr. Iizuka, Japan.30) Levan (from Erwinia herbicola) was purchased from Sigma Aldrich Co. (St. Louis, USA).
Measurement of enzyme activity. A reaction mixture consisting of 25 μL of enzyme, 50 μL of 200 mM nystose in distilled water, 25 μL of 100 mM sodium acetate buffer (pH 4.6), and a small amount of toluene was incubated at 30°C. The reaction was stopped by boiling for 3 min. One unit of 1-FEH activity was defined as the amount of enzyme required to produce 1 μmol of fructose per min under the above reaction conditions. For the analysis of the reaction products, HPAEC was performed using a DX500 chromatograph (Dionex Co., Sunnyvale, USA) with a CarboPac PA-1 anion exchange column (Dionex) and a pulsed amperometric detector (PAD) as described previously.29) The gradient was established by mixing eluent A (150 mM NaOH) with eluent B (500 mM acetate‒Na in 150 mM NaOH) in two ways. System I: 0‒1 min, 25 mM; 1‒2 min, 25‒50 mM; 2‒20 min, 50‒200 mM, 20‒22 min, 500 mM; 22‒30 min, 25 mM. System II: 0‒1 min, 25 mM; 1‒2 min, 25‒50 mM; 2‒14 min, 50‒500 mM, 14‒22 min, 500 mM; 22‒30 min, 25 mM. The flow rate through the column was 1.0 mL/min. The applied PAD potentials for E1 (400 ms), E2 (200 ms), and E3 (400 ms) were 0.05, 0.75 and -0.15 V, respectively, and the output range was 1 µC.
McIlvaine buffer (Na2HPO4-citric acid buffer, pH range 3.0‒8.0) was used for the estimation of the optimum pH. The reaction was stopped by the addition of 900 μL of 150 mM NaOH. For pH-stability profiles, a mixture of 25 μL of Britton Robinson buffer (NaOH-phosphoric acid, acetic acid, and boric acid buffer, pH range 3.0‒9.0) and 25 μL of enzyme solution (0.04 U/mL) was kept at 4°C for 24 h. The mixture was then adjusted to pH 4.6, and incubated with 100 mM nystose at 30°C for 30 min. The reaction was stopped in a boiling water bath for 3 min. For temperature stability profiles, enzyme solutions (0.04 U/mL) were pre-incubated with 100 mM sodium acetate buffer (pH 4.6) for 15 min at 4, 30, 40, 50, or 60°C. The pre-incubated solution was subsequently cooled to 0°C, and then the mixtures were incubated with nystose at 30°C for 30 min. All the experiments were performed in duplicate.
Quantitative determination of proteins. Proteins were determined by measuring their absorbance at 280 nm with reference to E1%1cm(extinction coefficient) = 10 as a tentative value.
Molecular cloning of 1-FEH cDNA from edible burdock. One gram of edible burdock root was ground in liquid nitrogen, and then was used for total RNA extraction by using an RNeasy Plant Mini Kit (Qiagen Inc., Valencia, USA). The first strand cDNA was synthesized using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen Co., Carlsbad, USA) to produce the templates for polymerase chain reaction (PCR) and rapid amplification of cDNA ends (RACE). Oligonucleotide sequences used for the cloning procedures are listed in Table 1. Degenerate PCR was done using Dp-E and Dp-F primers, which were designed according to the amino-acid sequences of FEHs from other plants. The PCR consisted of an initial 2 min denaturation step (94°C), followed by 35 cycles of 94°C for 30 s (denaturation), 50°C for 30 s (annealing) and 72°C for 1 min (extension), concluding with a final extension at 72°C for 7 min. The PCR was performed using Blend Taq polymerase (Toyobo Co., Osaka, Japan). The PCR product was ligated into pGEM-T vector (Promega Co., Madison, USA). The ligation product was transformed into Escherichia coli DH5α cells (Toyobo). The plasmid was purified by a GenElute Plasmid Mini-Prep Kit (Sigma Aldrich), and its insert DNA was sequenced. The insert was denoted as DegeEHb.

On the resultant DegeEHb DNA sequence, the gene-specific primers (5Ra-EHb1 and 3Ra-EHb1) were designed, and used for 5′ and 3′ RACE to amplify the 5′- and 3′-flanking regions of DegeEHb. RACE was performed using a Gene Racer Kit (Invitrogen) and KOD plus (Toyobo). The 5′ and 3′ RACE consisted of an initial 2 min denaturation step (94°C), followed by 40 cycles of 98°C for 10 s and 68°C for 50 s, and then a final step at 68°C for 10 min. Finally, the 5′ part of the cDNA was obtained by nested PCR with GeneRacer 5′Nested Primer and 5Ra-EHb2. Next, the 3′ part of the cDNA was obtained by nested PCR with GeneRacer 3′ Nested Primer and 3Ra-EHb2. These PCRs were performed under the same conditions as the first RACE PCR. After an adenine residue was attached to the RACE products with an A-addition kit (Qiagen), these products were subcloned into pGEM-T vector, and then its insert DNA was sequenced.
Gene-specific primers (EHb-Fw1 and EHb-Rv1) were designed from the DNA sequences of the 5′ and 3′ RACE products, and subjected to PCR to amplify the DNA corresponding to the open reading frame of the target gene. The PCR consisted of an initial 2 min denaturation step (94°C), followed by 35 cycles of 94°C for 15 s, 50°C for 30 s, and 68°C for 1.5 min, and then a final step at 68°C for 10 min. The amplified DNA fragments, named PsEHb were sequenced. Full length cDNA was compiled by overlapping the sequences of PsEHb and RACE PCR products. The full length cDNA was named aleh2. The nucleotide sequence of the full length cDNA has been submitted to GenBank, EMBL, and DDBJ (AB909128).
Expression of recombinant proteins in a methylotrophic yeast. The isolated cDNA aleh2 was expressed in the methylotrophic yeast Pichia pastoris with the secretory expression vector pPICZαB (EasySelect Pichia Expression Kit, Invitrogen). To construct expression plasmid, which was called pPic_aleh2, a DNA fragment containing a gene sequence of aleh2 corresponding to the mature protein was amplified by PCR using EHb-PstI and EHb-XbaI as primers. These primers also had recognition sequences for PstI and XbaI incorporated into forward and reverse primers, respectively (Table 1). The PCR condition was 1 cycle of 94°C for 2 min, 30 cycles of 94°C for 30 s, 52°C for 30 s and 68°C for 1.5 min, followed by 1 cycle of 68°C for 7 min using KOD plus and PsEHb as template DNA. The PCR products were digested with PstI and XbaI followed by ligation into a pPICZαB plasmid vector. The resulting plasmid pPic_aleh2 carries a Zeocin resistance gene and contains a gene sequence of aleh2 corresponding to the mature protein in frame behind the α-factor signal sequence. The pPic_aleh2 was sequenced to ensure no alteration of sequence in comparison with that of original aleh2.
Transformation and cultivation of P. pastoris were performed according to the instructions of the manufacturer with minor modifications. P. pastoris X-33 was transformed with 20 μg of the SacI-linearized vectors by electroporation, and the transformants were selected on YPDS (yeast extract pepton dextrose sorbitol)-Zeocin agar plates. Further production of recombinant protein was performed by the same procedures described in a previous study.17) To compare the properties of recombinant protein of aleh2 (rAlEH2), the purified recombinant protein of aleh1 (rAlEH1) was prepared by previously described method.17)
Purification of recombinant protein. All operations throughout the purification were performed at 0‒4°C. The recombinant protein was purified from the supernatant by DEAE Sepharose FF, HiTrap Phenyl FF, HiTrap Q HP, and Sephadex G-100 column chromatographies. The supernatant (450 mL) was dialyzed for 1 day against the 10 mM sodium phosphate buffer (pH 6.5). The dialyzate was applied to a column (φ3.0×25 cm) of DEAE-Sepharose FF (GE Healthcare Co., Buckinghamshire, UK) equilibrated with 10 mM sodium phosphate buffer (pH 6.0). The adsorbed proteins were eluted with a linear gradient of 0‒0.5 M sodium chloride in the same buffer. The active fraction was dialyzed overnight against 30% saturated ammonium sulfate in 10 mM sodium phosphate buffer (pH 6.0). The dialyzate was loaded on to a HiTrap Phenyl FF column (GE Healthcare) equilibrated with the same buffer. The elution was achieved with a linear gradient from 30% to 0% saturation of ammonium sulfate in the same buffer. Each active fraction was dialyzed overnight against 20 mM Bis-Tris buffer (pH 6.5). The dialyzate was applied to a HiTrap Q HP column (GE Healthcare) equilibrated with the same buffer. Each active fraction was dialyzed overnight against 50 mM sodium phosphate buffer containing 100 mM sodium chloride (pH 6.0). The dialyzate was concentrated to 1.0 mL by ultrafiltration on a VivaSpin 15R concentrator cutting off at 30 kDa (Vivascience Ltd., Stonehouse, UK). The concentrated solution was filtered on a column (φ1.8×67 cm) of Sephadex G-100 equilibrated with the same buffer.
Expression analysis of 1-FEH genes by quantitative RT-PCR. Total RNA was extracted from the burdock roots stored at 0 or 20°C for 0, 7, 14, 21, 28, and 42 days using an RNeasy Plant Mini Kit (Qiagen) with DNase I treatment (Qiagen) to remove genomic DNA. Each cDNA was synthesized from 500 ng of total RNA using a PrimeScript RT reagent Kit (Takara Bio Inc., Ohtsu, Japan) with oligo dT primer according to the manufacturer’s procedures. Obtained cDNAs were diluted 20-fold, and then used as templates for quantitative PCR using SYBR Premix ExTaq II (Tli RNaseH Plus) (Takara Bio). Transcript levels of target and reference genes were quantified by ABI 7300 Real Time PCR Systems (Applied Biosystems Inc., Foster City, USA). Dilutions of cDNAs prepared from burdock roots stored at 0°C for 14 days were used as the standard curve for relative quantification. The target genes were normalized with the value of the actin gene as a reference gene. To confirm the sequence of actin in burdock, the actin gene was amplified by PCR with primers UnACT_Fw (5′-TGGCATCACACTTTCTACAA-3′) and UnACT_Rv (5′-GTGAACATGTAACCTCTTTC-3′) which were designed according to the sequence of actin in Asteraceae plants such as Dahlia pinnata, Xanthium strumarium, Stevia rebaudiana, Helianthus annuus, and Chrysanthemum lavandulifolium (accession number: AB621922, JF434697, AF548026, FJ487620, JN638568). The nucleotide sequence of the putative actin gene has been submitted to GenBank, EMBL, and DDBJ (AB914771). A gene specific primer for quantitative PCR was constructed using Primer 3 software. Primer sequences are shown in Table 2. Thermal conditions of the quantitative PCR consisted of an initial 10 denaturation step (95°C), followed by 50 cycles of 95°C for 5 s, and 60°C for 30 s. Results were obtained from three individual roots and analyzed by SDS software ver. 1.4 (Applied Biosystems).

RESULTS AND DISCUSSION
Molecular cloning of edible burdock 1-FEH cDNA.
Edible burdock 1-FEH cDNA, which differed from the aleh1 already identified, was obtained by the RACE method following PCR using degenerate primers based on FEH amino-acid sequences from other plants (Table 1). A full-length cDNA, named aleh2, consisted of 1,817 bp and contained an open reading frame (ORF) of 1,701 bp and a poly (A) sequence at the 3′ end. The ORF encoded a polypeptide of 566 amino acids (Fig. 1). The deduced polypeptide was denoted as AlEH2. The primary sequence of AlEH2 exhibits great similarity with that of chicory 1-FEH I (78%), but only 52% similarity with chicory 1-FEH IIa/IIb and AlEH1. Phylogenetic tree analysis, based on the deduced amino acid sequences, showed that AlEH2 is present in a group of cell wall invertases, as well as other FEHs (data not shown). The molecular mass and isoelectric point (pI) of AlEH2 were calculated to be 63,907 and 5.55, respectively. The amino acid sequence contains ten putative N-glycosylation sites (N-X-S/T). AlEH2 contains important amino acid residues (Asp55, Asp180, Glu234) which are conserved in the GH32 family.22) The role of the amino acid residues of GH32 was identified by Reddy and Maley31) 32) and by Meng and Fütterer.33)

Heterologous expression and purification of recombinant protein.
A heterologous expression system using Pichia pastoris is highly suitable for producing the translation products from the genes of plant fructosyltransferases and invertases.34) P. pastoris does not produce fructosyltransferases or invertases; background activities of these enzymes were not detected in the culture medium of P. pastoris. A recombinant protein was obtained by the expression of aleh2 in P. pastoris. From the enzyme assay with nystose as a substrate, hydrolytic activity was detected in the culture medium of P. pastoris. The enzyme was partially purified from the culture medium by ion exchange chromatography with DEAE-Sepharose FF, HiTrap Phenyl FF, HiTrap Q HP and Sephadex G-100. The active fraction separated by gel filtration chromatography, was used as a partially purified enzyme. Purification procedures used to obtain the recombinant protein are summarized in Table 3. The specific activity of the recombinant protein was 10.8 U/mg protein.

General properties of the enzyme.
The optimum pH of the recombinant AlEH2 was 4.6. The enzyme was stable in the range of pH 5.8‒7.2 at 4°C for 24 h (residual activity more than 80%). The enzyme was stable up to 30°C (residual activity more than 80%), and inactivated at 50°C in pre-incubation in pH 4.6 for 15 min.
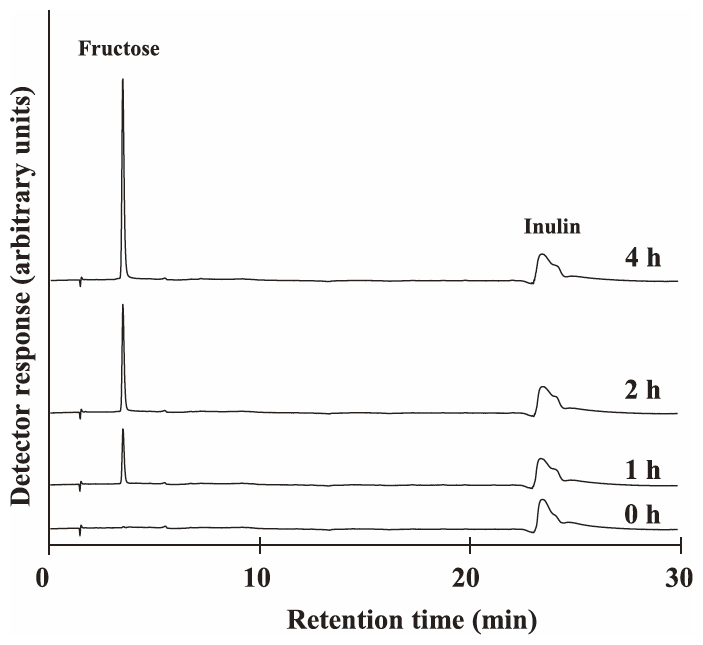
Recombinant protein rAlEH2 was incubated in 10 mM inulin (MW 5,000) at 30°C for 0, 1, 2, and 4 h. Inulin was hydrolyzed by this enzyme and fructose was produced (Fig. 2). Increases in the peaks of IOS, such as inulotriose (F3) and inulotetraose (F4), were not observed in the reaction mixture. Therefore, it was confirmed that rAlEH2 was not endo-inulinase, which produced IOS from inulin.
Sucrose, neokestose, 6-kestose, and bacterial levan were hardly hydrolyzed (Table 4). Therefore, the enzyme is neither invertase nor 6-FEH. High hydrolase activity was exhibit against inulin-type fructans such as 1-kestose, nystose, fructosylnystose, or inulin. rAlEH2 hydrolyzed nystose, fructosylnystose, and inulin faster than 1-kestose, whereas the hydrolase activity of rAlEH1 showed the opposite results (Table 4). These results suggest that rAlEH2 is a 1-FEH enzyme that efficiently hydrolyzes fructans [1F(1-β-D-fructofuranosyl)m sucrose, m = 1~ca.30], which have DP higher than 1-kestose, whereas rAlEH1 is a 1-FEH that efficiently hydrolyzes 1-kestose.

To elucidate whether sucrose acts as inhibitor on these recombinant proteins, enzymatic activity was assayed in 10 mM nystose with and without sucrose (Fig. 3). Sucrose inhibited the enzymatic activity of rAlEH1 but not of rAlEH2. These results were in agreement with the properties of chicory 1-FEH I and 1-FEH IIa/b. In chicory 1-FEH IIa, W82 and S101 have been identified as important amino acid residues for binding sucrose as inhibitor.35) The positions of these amino acid residues in AlEH1 are identical, and those in AlEH2 are leucine and glutamine the same as chicory 1-FEH I (Fig. 1). From these findings, the rAlEH2 was the 1-FEH that is not inhibited by sucrose.
Gene expression of 1-FEHs in burdock roots stored in low temperature.
Expression levels of aleh1 and aleh2 in burdock roots stored at 0 or 20°C were measured by real time RT-PCR (Fig. 4). The expression level of aleh2 was shown to be as high as that of aleh1 in roots during low-temperature storage from 14 days to 28 days. However, this drastic change in the expression levels of both genes was not found in burdock stored at 20°C (Fig. 4). The highest expression levels were detected at 0°C, with aleh1 peaking after 14 days of storage, and aleh2 peaking after 21 days of storage. These results suggest that the two 1-FEH genes are induced by low temperature. Maroufi et al.36) have reported on the expression levels of the 1-FEH genes in chicory roots (three cultivars). The expression levels of 1-FEH I gene are lower than the expression levels of 1-FEH II gene in chicory roots stored at 4°C. Chicory 1-FEH genes seem to exhibit the differential expression profiles in chicory roots stored cold condition for five and nine weeks. In case of burdock stored at 0°C until six weeks (42 days), expression pattern of aleh2, which is similar to chicory 1-FEH I gene, was correspondingly displayed that of aleh1, which is similar to chicory 1-FEH II gene. We need to analyze the expression patterns of FEH genes in burdock during longer time storage at 0 or 4°C in further work.

We have previously reported on the variation in the enzyme activity of burdock roots stored at different temperatures, and we observed that the 1-kestose hydrolyzing activity in the roots after 14 days of storage at 0°C was higher than that in the roots before storage.10) The pattern of the 1-kestose hydrolyzing enzyme activity in burdock during the low-temperature storage is similar to that of the expression level of aleh1. However, inulinase activity decreased gradually with the storage time in spite of the observation that aleh1 and aleh2 were highly expressed in roots after 21 days and 28 days of storage. The reason for this difference is still not clear and requires further study, including the expression analysis of protein levels using western blotting or a more detailed measurement of enzyme activities.
In this study, we cloned and characterized a cDNA, called aleh2, which encodes for the edible burdock 1-FEH. Recombinant protein of aleh2 was expressed in Pichia pastoris and partially purified. The recombinant protein of aleh2 showed 1-FEH activity that released fructose from 1-kestose, nystose, fructosylnystose, and inulin.
Unlike the aleh1 recombinant protein previously reported, this enzyme preferred to hydrolyze longer-chain fructans than 1-kestose. Through expression analysis of aleh1 and aleh2, high expression levels of these genes were observed in the burdock roots stored at 0°C. Thus, we confirmed that the genes of two 1-FEHs, aleh1 and aleh2, are involved in the hydrolysis of fructans in burdock roots during low temperature storage.
ACKNOWLEDGMENTS
This work was partially supported by a Grant-in-Aid to Cooperative Research from Rakuno Gakuen University. Rakuno Gakuen University Dairy Science Institute, 2011‒3. We thank Dr. Masaru Iizuka, Kobe Shoin Women’s University, for providing 6-kestose.
REFERRENCES
- 1 J. Edelman and T. Jefford: The mechanism of fructosan metabolisim in higher plants as exemplified in Helianthus tuberosus. New Phytol., 67, 517-531 (1968).
- 2) N. Shiomi and M. Izawa: Purification and characterization of sucrose:sucrose 1-fructosyltransferase from the roots of asparagus (Asparagus officinalis L.). Agric. Biol. Chem., 44, 603-614 (1980).
- 3) A.J. Koops and H.H. Jonker: Purification and characterization of the enzymes of fructan biosynthesis in tubers of Helianthus tuberosus Colombia. II. Purification of sucrose:sucrose 1-fructosyltransferase and reconstitution of fructan synthesis in vitro with purified sucrose:sucrose 1-fructosyltransferase and fructan:fructan 1-fructosyltransferase. Plant Physiol., 110, 1167-1175 (1996).
- 4) M. Lüscher, C. Erdin, N. Sprenger, U. Hochstrasser, T. Boller, and A. Wiemken: Inulin synthesis by a combination of purified fructosyltransferases from tubers of Helianthus tuberosus. FEBS Lett., 385, 39-42 (1996).
- 5) N. Shiomi: Purification and characterisation of 1F-fructosyltransferase from the roots of asparagus (Asparagus officinalis L.). Carbohydr. Res., 99, 157-169 (1982).
- 6) A.J. Koops and H.H. Jonker: Purification and characterization of the enzymes of fructan biosynthesis in tubers of Helianthus tuberosus ‘Colombia’. I. Fructan:fructan fructosyltransferase. J. Exp. Bot., 45, 1623-1631 (1994).
- 7) W. Van den Ende, D. Van Wonterghem, P. Verhaert, E. Dewil, and A. Van Laere: Purification and characterization of fructan:fructan fructosyltransferase from chicory roots (Cichorium intybus L.). Planta, 199, 493-502 (1996).
- 8) M. Abe, K. Ueno, Y. Ishiguro, T. Omori, S. Onodera, and N. Shiomi: Purification, cloning and functional characterization of fructan: fructan 1-fructosyltransferase from edible burdock (Arctium lappa L.). J. Appl. Glycosci., 56, 239-246 (2009).
- 9) Y. Ishiguro, K. Ueno, M. Abe, S. Onodera, E. Fukushi, N. Benkeblia, and N. Shiomi: Isolation and structural determination of reducing fructooligosaccharides newly produced in stored edible burdock. J. Appl. Glycosci., 56, 159-164 (2009).
- 10) Y. Ishiguro, S. Onodera, N. Benkeblia, and N. Shiomi: Variation of total FOS, total IOS, inulin and their related-metabolizing enzymes in burdock roots (Arctium lappa L.) stored under different temperatures. Postharvest Biol. Technol., 56, 232-238 (2010).
- 11) Y. Ishiguro, K. Ueno, S. Onodera, N. Benkeblia, and N. Shiomi: Effect of temperatures on inulobiose and inulooligosaccharides in burdock roots during storage. J. Food Compost. Anal., 24, 398-401 (2011).
- 12) W. Van den Ende, J. De Roover, and A. Van Laere: In vitro synthesis of fractofuranosyl-only oligosaccharides from inulin and fructose by purified chicory root fructan:fructan fructosyl transferase. Physiol. Plant., 97, 346-352 (1996).
- 13) W. Saengthongpinit and T. Sajjaanantaku: Influence of harvest time and storage temperature on characteristics of inulin from Jerusalem artichoke (Helianthus tuberosus L.) tubers. Postharvest Biol. Technol., 37, 93-100 (2005).
- 14) W. Van den Ende, A. Michiels, J. De Roover, P. Verhaert, and A. Van Laere: Cloning and functional analysis of chicory root fructan 1-exohydrolase I (1-FEH I): a vacuolar enzyme derived from a cell-wall invertase ancestor? Mass fingerprint of the 1-FEH I enzyme. Plant J., 24, 447-456 (2000).
- 15) W. Van den Ende, A. Michiels, D. Van Wonterghem, S. Clerens, J. De Roover, and A. Van Laere: Defoliation induces 1-FEH II (fructan 1-exohydrolase II) in witloof chicory roots: cloning and purification of two isoforms (1-FEH IIa and 1-FEH IIb). Plant Physiol., 126, 1186-1195 (2001).
- 16) W. Van den Ende, S. Clerens, R. Vergauwen, L. Van Riet, A. Van Laere, M. Yoshida, and A. Kawakami: Fructan 1-exohydrolases: β (2,1) trimmers during graminan biosynthesis in stems of wheat? Purification, characterization, mass mapping and cloning of two fructan 1-exohydrolase isoforms. Plant Physiol., 131, 621-631 (2003).
- 17) K. Ueno, Y. Ishiguro, M. Yoshida, S. Onodera, and N. Shiomi: Cloning and functional characterization of a fructan 1-exohydrolase (1-FEH) in edible burdock (Arctium lappa L.). Chem. Cent. J., 5, 16 (2011).
- 18) J. Lothier, B. Lasseur, K. Le Roy, A. Van Laere, M.P. Prud’homme, P. Barre, W. Van den Ende, and A. Morvan-Bertrand: Cloning, gene mapping, and functional analysis of a fructan 1-exohydrolase (1-FEH) from Lolium perenne implicated in fructan synthesis rather than in fructan mobilization. J. Exp. Bot., 58, 1969-1983 (2007).
- 19) L. Van Riet, V.J. Nagaraj, W. Van den Ende, S. Clerens, A. Wiemken, and A. Van Laere: Purification, cloning and functional characterization of a fructan 6-exohydrolase from wheat (Triticum aestivum L.). J. Exp. Bot., 57, 213-223 (2006).
- 20) A. Kawakami, M. Yoshida, and W. Van den Ende: Molecular cloning and functional analysis of a novel 6&1-FEH from wheat (Triticum aestivum L.) preferentially degrading small graminans like bifurcose. Gene, 358, 93-101 (2005).
- 21) W. Van den Ende, A. Michiels, J. De Roover, and A. Van Laere: Fructan biosynthetic and breakdown enzymes in dicots evolved from different invertases. Expression of fructan genes throughout chicory development. Sci. World J., 2, 1281-1295 (2002).
- 22) B. Henrissat: A classification of glycosyl hydrolases based on amino-acid sequence similarities. Biochem. J., 280, 309-316 (1991).
- 23) M. Verhaest, W. Van den Ende, K. Le Roy, C.J. De Ranter, A. Van Laere, and A. Rabijns: X-ray diffraction structure of a plant glycosyl hydrolase family 32 protein: fructan 1-exohydrolase IIa of Cichorium intybus. Plant J., 41, 400-411 (2005).
- 24) A. Michiels, A. Van Laere, W. Van den Ende, and M. Tucker: Expression analysis of a chicory fructan 1-exohydrolase gene reveals complex regulation by cold. J. Exp. Bot., 55, 1325-33 (2004).
- 25) A.F. Asega, J.R. do Nascimento, and M.A. Carvalho: Increased expression of fructan 1-exohydrolase in rhizophores of Vernonia herbacea during sprouting and exposure to low temperature. J. Plant Physiol., 168, 558-565 (2011).
- 26) A.F. Asega, J.R. do Nascimento, L. Schroeven, W. Van den Ende, and M.A. Carvalho: Cloning, characterization and functional analysis of a 1-FEH cDNA from Vernonia herbacea (Vell.) Rusby. Plant Cell Physiol., 49, 1185-1195 (2008).
- 27) F. del Viso, A.F. Puebla, H.E. Hopp, and R.A. Heinz: Cloning and functional characterization of a fructan 1-exohydrolase (1-FEH) in the cold tolerant Patagonian species Bromus pictus. Planta, 231, 13-25 (2009).
- 28) H. Takeda, K. Sato, S. Kinoshita, and H. Sasaki: Production of 1-kestose by Scopulariopsis brevicaulis. J. Ferment. Bioeng., 77, 386-389 (1994).
- 29) N. Shiomi: Structure of fructopolysaccharide (asparagosin) from roots of asparagus (Asparagus officinalis L.). New Phytol., 123, 263-270 (1993).
- 30) M. Iizuka, H. Yamaguchi, S. Ono, and N. Minamiura: Production and isolation of levan by use of levansucrase immobilized on the ceramic support SM-10. Biosci. Biotechnol. Biochem., 57, 322-324 (1993).
- 31) V.A. Reddy and F. Maley: Identification of an active-site residue in yeast invertase by affinity labeling and site-directed mutagenesis. J. Biol. Chem., 265, 10817-10820 (1990).
- 32) A. Reddy and F. Maley: Studies on identifying the catalytic role of Glu-204 in the active site of yeast invertase. J. Biol. Chem., 271, 13953-13957 (1996).
- 33) G. Meng and K. Fütterer: Structural framework of fructosyl transfer in Bacillus subtilis levansucrase. Nat. Struct. Biol., 10, 935-941 (2003).
- 34) A. Kawakami and M. Yoshida: Molecular characterization of sucrose:sucrose 1-fructosyltransferase and sucrose:fructan 6-fructosyltransferase associated with fructan accumulation in winter wheat during cold hardening. Biosci. Biotechnol. Biochem., 66, 2297-2305 (2002).
- 35) M. Verhaest, W. Lammens, K. Le Roy, C. J. De Ranter, A. Van Laere, A. Rabijns, and W. Van den Ende: Insights into the fine architecture of the active site of chicory fructan 1-exohydrolase: 1-kestose as substrate vs sucrose as inhibitor. New Phytol., 174, 90-100 (2007).
- 36) A. Maroufi, E. Van Bockstaele, and M. De Loose: Differential expression of fructan 1-exohydrolase genes involved in inulin biodegradation in chicory (Chicorium intybus) cultivars. Aust. J. Crop Sci., 6, 1362-1368 (2012).

