Abstract
The cellulose binding domain (CBD) of cellulosome-integrating protein A from Clostridium thermocellum NBRC 103400 was genetically fused to FMN-dependent NADH-azoreductase (AZR) and glucose 1-dehydrogenase (GDH) from Bacillus subtilis. The fusion enzymes, AZR-CBD and CBD-GDH, were expressed in Escherichia coli Rosetta-gami B (DE3). The enzymes were purified from cell-free extracts, and the specific activity of AZR-CBD was 15.1 U/mg and that of CBD-GDH was 22.6 U/mg. AZR-CBD and CBD-GDH bound strongly to 0.5 % swollen cellulose at approximately 95 and 98 % of the initial protein amounts, respectively. After immobilization onto the swollen cellulose, AZR-CBD and CBD-GDH retained their catalytic activity. Both enzymes bound weakly to 0.5 % microcrystalline cellulose, but the addition of a high concentration of microcrystalline cellulose (10 %) improved the binding rate of both enzymes. A reactor for flow injection analysis was filled with microcrystalline cellulose-immobilized AZR-CBD and CBD-GDH. This flow injection analysis system was successfully applied for the determination of glucose, and a linear calibration curve was observed in the range of approximately 0.16–2.5 mM glucose, with a correlation coefficient, r, of 0.998.
Abbreviations
CBD, cellulose binding domain; AZR, FMN-dependent NADH-azoreductase; GDH, glucose 1-dehydrogenase; DCIP, 2,6-dichloroindophenol; FMN, flavin mononucleotide; NAD(H), β-nicotinamide adenine dinucleotide; Cip A, cellulosome-integrating protein A; CBM, carbohydrate-binding module; IPTG, Isopropyl β-D-1-thiogalactopyranoside; SDS-PAGE, Sodium dodecyl sulfate polyacrylamide gel electrophoresis.
INTRODUCTION
A number of oxidases and dehydrogenases with high substrate specificity have been used for analytically purposes.1) They can specifically detect sugars, acids, alcohols, and other minor components in food, feed, and blood. These reactions are easily monitored using photometric,2) fluorimetric,3) and electrometric analytical methods.4) As a consequence, colorimetric assay reagent kits and portable electric sensors have become available commercially. In addition, these enzymes are also used in high-precision laboratory instruments, such as electrochemical analyzers, optical biosensors, and flow injection systems. For biosensor applications, types of oxidoreductase may be immobilized on the electrode, sensor chip, and resin beads via supports and matrixes. In many cases, enzyme immobilization employs absorption, covalent binding, or chemical cross-linking methods.5) The absorption method is simple, but the binding capacity of the enzyme is very low. In the cases of covalent binding and chemical cross-linking method, supports and matrixes strongly retain the enzymes. However, the procedures for immobilization are complicated, and sometimes the activity of enzyme is reduced or completely lost.
Construction of recombinant enzymes with binding ability to supports and matrixes is one way to improve enzyme immobilization. From this point of view, we focused cellulose binding domains (CBDs). CBDs have been found in cellulases and cellulosomes, and the biochemical properties of CBDs have been investigated.6)7)8) Moreover, some CBD-fusion recombinant enzymes have been reported,9)10) and immobilization of the fusion enzyme was successful.11)12) Therefore, fusion of CBD to the oxidoreductase allows the recombinant enzyme to be immobilized on cellulosic supports for biosensor production without complicated processing.
In this study, we selected FMN-dependent NADH-azoreductase (AZR, accession No. NP_391234) and glucose 1-dehydrogenase (GDH, NP_388275) of Bacillus subtilis 168,13)14) as a model oxidoreductase for the biosensor and a recipient of CBD fusion. AZR is an FMN-linked enzyme and catalyzes the reduction of dichloroindophenol (DCIP) using NADH as an electron donor. Because AZR can decolorize DCIP by reduction, AZR is useful for colorimetric determination of NADH. GDH catalyzes the oxidation of D-glucose to D-glucono-1,5-lactone and the reduction of NAD+ to NADH. Therefore, GDH is a model enzyme for NAD+ reductases in this study. By combining the AZR and GDH reactions, glucose was detected as DCIP reductant by spectrophotometry (Fig. 1A). As a model of CBD, we selected the CBD from cellulosome-integrating protein A (Cip A, YP_001039466) of Clostridium thermocellum NBRC 103400, which is classified into carbohydrate-binding module family 3 (CBM3),15) on the basis of their amino acid sequences referenced to the Carbohydrate Active enZymes (CAZy) database (http://www.cazy.org/). The binding site of CBD comprises a planar hydrophobic platform, which contains exposed aromatic amino acids, and the binding site interacts with the flat surfaces of cellulose.16)
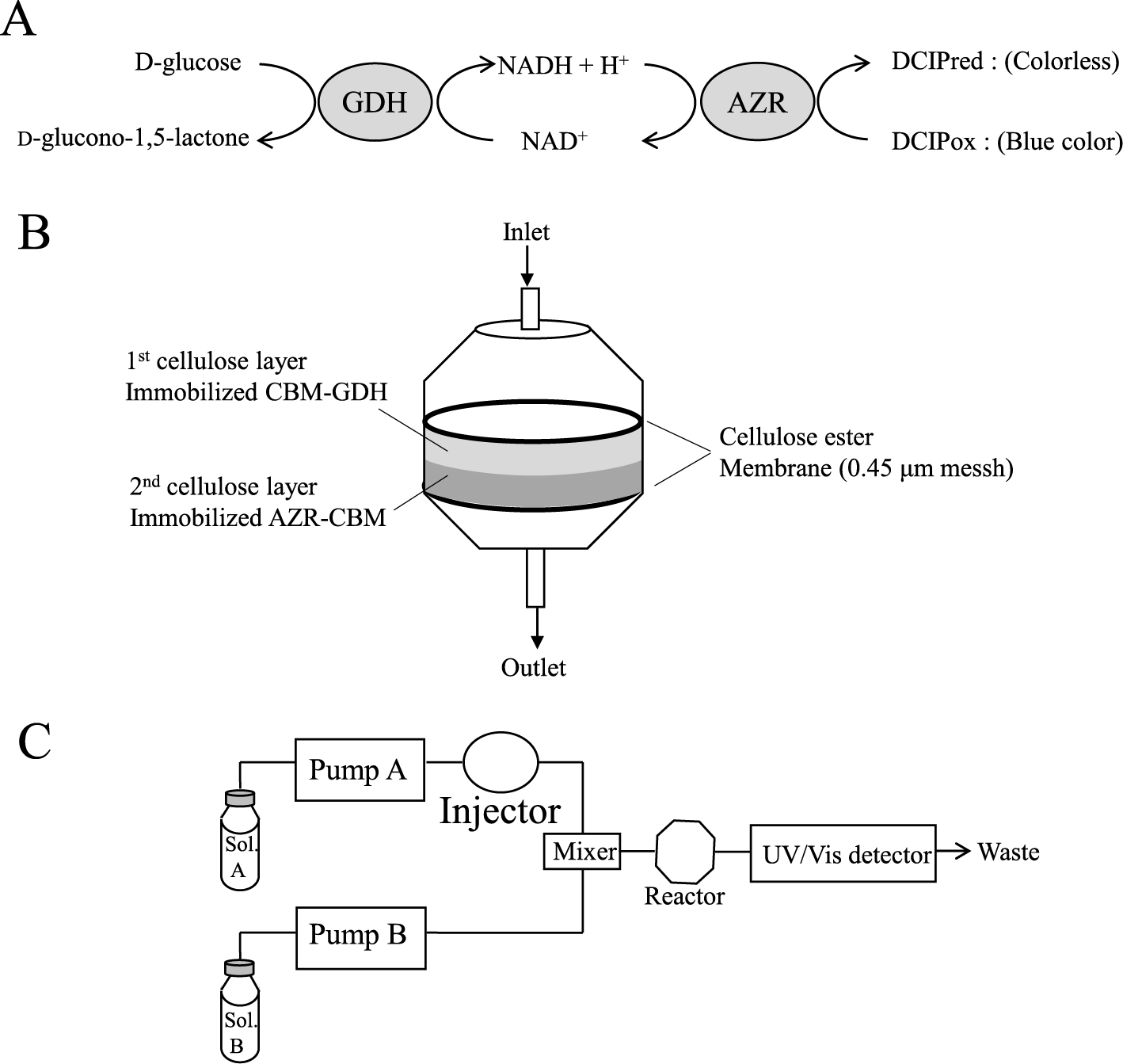
Fig. 1.
Schematic diagram of the reaction mechanism for the detection of glucose (A), reactor for flow injection analysis (B), and flow injection analysis system for the determination of glucose.
Considering these findings, we constructed CBD-fusion AZR and GDH via chimeric genes and expressed them in E. coli. To immobilize the fusion enzymes, the binding abilities were analyzed using swollen cellulose and microcrystalline cellulose. Flow injection analysis system for the determination of glucose was developed using a reaction column consisting the fusion enzymes immobilized on cellulose.
MATERIALS AND METHODS
Microorganisms and culture. Escherichia coli DH5α cells (TOYOBO, Osaka, Japan) used as a host to construct various recombinant plasmids were grown at 37 °C with shaking (100 rpm) in LB medium containing 100 μg/mL ampicillin. E. coli Rosetta-gami B (DE3) (Novagen, Madison, WI, USA), which harbored a recombinant plasmid, was grown at 30 °C with shaking (100 rpm) in LB medium containing 100 μg/mL ampicillin, 10 μg/mL chloramphenicol, 25 μg/mL kanamycin, and 15 μg/mL tetracycline.
Expression of wild-type and CBD-fusion enzyme genes. All primers for PCR are listed in Table 1. The AZR (gene symbol; yvaB) and GDH (gene symbol; gdh) genes were amplified from genomic DNA of Bacillus subtilis 168 by PCR. The primer pairs for amplifying the DNA fragments with AZR and GDH were bsAZR-Nde/bsAZR-Xho and FBSgdhNde1/BSgdhXho1R, respectively. The PCR fragments were digested with NdeI and XhoI, and then cloned into the NdeI and XhoI sites of pET22b (+). The recombinant plasmids were designated pET22b-azr and pET22b-gdh, respectively, and both plasmids were used for the expression of wild-type enzymes.
Table 1.
Oligonucleotide primers
| Primer name |
Oligonucleotide sequence |
| bs AZR-Nde |
5′-CGAAAGGGATGGTACATATGGCAAAAG -3′ |
| bsAZR-Xho |
5′- CCAAATAACTCGAGGCTTTTTCATTAG-3′ |
| FBSgdhNde1 |
5′-GGAGGAGGATGCATATGTATCCGGATT -3′ |
| BSgdhXho1R |
5′- ATGTCTGGGTCGCTCGAGATGTTTAACCG-3′ |
| CBDBamF |
5′- CATTTATGGATCCTGGTGTTAACGTTGGC-3′ |
| CBDXhoR |
5′- GTTACAGGCTCGAGTGATGGTACTACACTG-3′ |
| bsAZR-BamR |
5′-GGCTGGATCCAAGAATGTTTTTCCTGCTTC-3′ |
| CBDNdeF |
5′-ACACCGACAAACCATATGGCAAATACACCG -3′ |
| CBDBamR |
5′-TGCATTCGGATCCTGACGGCGGTATTGT-3′ |
| GDHBamF |
5′-TAAGAAGGAGATGGATCCATGTATCCGGAT-3′ |
| GDHBam2 |
5′-ATAGGATCCGGAGGAGGAGGTGGTGGTATGTAT-3′ |
| T7 promoter |
5′- CCCGCGAAATTAATACGACTCACTATAGGG-3′ |
| T7 terminator |
5′- CAAGGGGTTATGCTAGTTATTGCTCAGCGG3′ |
Restriction sites in the oligonucleotide sequence are underlined.
To construct expression plasmids for AZR- and CBD-fusion enzymes, the TP linker and CBD gene were amplified from genomic DNA of C. thermocellum NBRC 103400 using the primer pair CBDBamF/CBDXhoR. After digesting the amplified fragment with BamHI and XhoI, the fragment was cloned into the BamHI and XhoI sites of pET22b(+) (Novagen), and the recombinant plasmid was designated pET22b-cbdC. The AZR gene was amplified by PCR with plasmid pET22b-azr as the template, and a primer pair for the T7 promoter and bsAZR-BamR was used. The PCR fragments were digested with NdeI and BamHI, and then cloned into the NdeI and BamHI sites of pET22b-cbdC. The resulting plasmid (pET22b-azr-cbd) encoded the fusion protein, AZR-CBD, which consisted of the N-terminal region of AZR, TP linker, CBD, and a C-terminal 6×His tag.
The CBD and GDH fusion enzyme (CBD-GDH) was designed as the N-terminal region of CBD, TP linker, Gly×6 repeats linker, and C-terminal GDH. The Gly×6 repeats linker was added to enhance the flexibility of the N-terminus and C-terminus. CBD and the TP linker gene were amplified from DNA of C. thermocellum using the primer pair CBDNdeF/CBDBamR. After digesting with NdeI and BamHI, the PCR fragment was cloned into the NdeI and BamHI sites of pET22b(+), and the recombinant plasmid was designated pET22b-Ncbd. To amplify the GDH gene and add a Gly×6 repeats linker sequence to the 5´ end of the GDH gene, two-round PCR was performed. In the first PCR, the pET22b-gdh plasmid was used as the template and a primer pair for the GDHBamF and the T7 terminator. In the second PCR, the PCR product from the first cycle was used as the template and the primer pair fGDHBam2/BSgdhXho1R. After digesting the amplified fragment with BamHI and XhoI, the fragment was cloned into the BamHI and XhoI sites of pET22b-Ncbd. The recombinant plasmid was designated pET22b-cbd-gdh.
All the expression plasmids were transfected into E. coli Rosetta-gami B (DE3) cells. The transformants were cultured at 30 °C using the conditions described above until the optical density at 600 nm was about 0.6, after which IPTG was added to the culture medium at a final concentration of 0.4 mM. The cultures were incubated at 30 °C for an additional 12 h.
Purification of recombinant enzymes. All procedures were performed at 0–4 °C, unless otherwise stated. E. coli cells harboring pET22b-azr-cbd, pET22b-azr, pET22b-cbd-gdh, or pET22b-gdh were each harvested from 500 mL of culture medium by centrifugation (10,000 × G for 10 min). Cells harboring pET22b-azr or pET22b-azr-cbd were suspended in 20 mL of 10 mM Tris-HCl buffer (pH 8.0), and those harboring pET22b-gdh or pET22b-cbd-gdh were suspended in 20 mL of 10 mM potassium phosphate buffer (pH 6.5) containing 10 % glycerol. Each of the cell suspensions was sonicated (4 °C, 10 min, 350–400 μA) on ice, after which cell debris was removed by centrifugation. The supernatant were dialyzed against the same buffer used for cell resuspension.
AZR-CBD was purified from the dialysate (cell-free extract) using a Ni-NTA column (2 × 3 cm; Ni-Sepharose 6 Fast Flow; GE Healthcare, Tokyo, Japan). The cell-free extract was applied to an Ni-NTA column equilibrated with 10 mM Tris-HCl (pH 8.0). After it was washed with the same buffer, the column was developed with buffer containing 100 mM imidazole, which eluted AZR-CBD. To purify AZR, the cell-free extract was applied to a DEAE-cellulofine A-500 column (3 × 5 cm; JNC Co., Tokyo, Japan) equilibrated with 10 mM Tris-HCl (pH 8.0), and the column was washed with this buffer. After washing with buffer, the column was developed with buffers containing 100 mM and 150 mM NaCl. AZR was eluted with buffer containing 150 mM NaCl, and the active fractions were dialyzed against buffer. Ammonium sulfate was added at 20 % saturation to the dialysate, and this solution was applied to a Butyl-Toyopearl 650M column (2 × 3 cm; TOSOH Co., Tokyo, Japan) equilibrated with 10 mM Tris-HCl (pH 8.0) containing 30 % ammonium sulfate. After washing with buffer containing 30 % ammonium sulfate, the column was developed with buffer containing 15 %, which eluted the enzyme.
Cell-free extract containing CBD-GDH was applied to a DEAE-cellulofine column (3 × 5 cm) equilibrated with 10 mM potassium phosphate buffer (pH 6.5) containing 10 % glycerol, and the column was washed with this buffer. After washing, the column was developed with buffers containing 20 mM and 60 mM NaCl. CBD-GDH was eluted with buffer containing 60 mM NaCl, and the active fractions were dialyzed against buffer. Ammonium sulfate was added at 10 % saturation to the dialysate, and this solution was applied to a Butyl-Toyopearl 650M column (2 × 3 cm) equilibrated with the buffer containing 10 % ammonium sulfate. After washing with the buffer containing 10 % ammonium sulfate, the column was developed with buffers containing 5 %, which eluted the enzyme. The cell-free extract containing GDH was applied to a DEAE-cellulofine column (3 × 5 cm) equilibrated with 10 mM potassium phosphate buffer (pH 6.5) containing 10 % glycerol, and the column was washed with this buffer. After washing, the column was developed with buffers containing 50 mM and 100 mM NaCl. GDH was eluted with buffer containing 100 mM NaCl, and the active fractions were dialyzed against buffer. Ammonium sulfate was added at 10 % saturation to the dialysate, and this solution was applied to a Butyl-Toyopearl 650M column (2 × 3 cm) equilibrated with buffer containing 10 % ammonium sulfate. After washing with buffer containing 10 % ammonium sulfate, and GDH was found in the unadsorbed fractions.
Assay for AZR activity. AZR activity was determined spectrophotometrically by measuring the reduction of DCIP at 600 nm. The mixture contained 40 μM DCIP, 0.1 mM NADH, 25 mM Tris-HCl buffer (pH 8.5), and appropriate amounts of the enzyme at 30 °C. One unit of enzyme activity was defined as the amount of enzyme catalyzing the reduction of 1 μmol of DCIP per minute.
Assay for GDH activity. GDH activity was determined spectrophotometrically by measuring the rate of NADH formation at 340 nm. The mixture contained 10 mM glucose, 1.0 mM NAD+, 25 mM potassium phosphate buffer (pH 6.5), and appropriate amounts of the enzyme at 30 °C. One unit of enzyme activity was defined as the amount of enzyme catalyzing the reduction of 1 μmol of NAD+ per minute.
Assay for cellulose binding activity. A mixture contained 0.5–10 % substrate, appropriate amounts of the enzyme, and 25 mM buffer. Tris-HCl buffer (pH 8.0) was used to determine the binding activity of AZR-CBD (0.55 mg/mL) and AZR (0.25 mg/mL), and potassium phosphate buffer (pH 6.5) was used to determine that of CBD-GDH (0.25 mg/mL) and GDH (0.13 mg/mL). After incubation for 10 min on ice with occasional stirring, the mixture was centrifuged, and the enzyme activity in the supernatant was determined. The amount of bound enzyme was estimated by subtracting the enzyme activity in the supernatant from the initial enzyme activity. The cellulose binding efficiency was given as the percentage of the initial enzyme activity. The values were averages for two independent experiments.
Preparation of the immobilized-enzyme reactor. A schematic diagram of immobilized-enzyme reactor column is shown in Fig. 1B. A stainless filter folder (inner diameter 8.0 mm; ADVANTEC, Tokyo, Japan) was used as a reactor column. The same volume of AZR-CBD (1.1 mg/mL) and microcrystalline cellulose solution (20 % w/v) containing 50 mM Tris-HCl buffer (pH 8.0) were mixed, and the mixture was incubated for 10 min on ice with occasional stirring. The mixture (100 μL) was filtered using a cellulose acetate filter (pore size 0.45 μm) on the stainless filter folder. CBD-GDH (0.5 mg/mL) was also mixed with microcrystalline cellulose solution (20 % w/v) containing 50 mM potassium phosphate buffer (pH 6.5), and the mixture was incubated for 10 min on ice with occasional stirring. The mixture (100 μL) was filtered, and cellulose-immobilized AZR-CBD was overlaid by cellulose immobilized CBD-GDH. After filtration, the cellulose acetate filter was put on the packed cellulose, and the reactor column was washed with 50 mM potassium phosphate buffer (pH 6.5).
Flow injection analysis system. A schematic diagram of flow injection analysis system is shown in Fig. 1C. As mobile phases, deionized water and 0.1 M potassium phosphate buffer (pH 6.5) containing 80 μM DCIP oxidant and 1 mM NAD+, were used and were pumped at a flow rate of 0.25 mL/min with LC-10AS pumps (Shimadzu Co., Kyoto, Japan). Glucose samples (50 μL) were injected into the deionized water line with a Rheodyne 7725 sample injector (IDEX, Rohner Park, CA, USA). The column was incubated at 30 °C using a column incubator. The formation of DCIP reductant was detected spectrophotometrically at 600 nm using an SPD-10A UV-Vis detector (Shimadzu). The activity of CBD-GDH was lost at pH 8. The potassium phosphate buffer (pH 6.5) was chosen to prevent inactivation of CBD-GDH.
Analytical methods. Protein concentrations were determined using the Lowry’s method with bovine serum albumin as the standard.17) In column chromatography, protein concentrations were followed using the absorbance at 280 nm. SDS-PAGE was performed in accordance with the Laemmli method.18) Pre-stained Protein Markers Broad Range (Nacalai Tesque Inc., Kyoto, Japan) and ExcelBand All Blue Regular Range Protein Marker PM1500 (SMOBIO Technology, Inc., Hsinchu, Taiwan) were used as a molecular marker.
Materials. Swollen cellulose was prepared in accordance with the methods described by Zhang et al. with slight modifications.19) Microcrystalline cellulose (0.2 g) was suspended in 0.6 mL of distilled water, and 10 mL of 86.2 % phosphoric acid was added to the cellulose mixture. The mixture was stirred until the cellulose was completely solubilized. Subsequently, 40 mL of ice-cold water was added and mixed well. The suspension was centrifuged at 8,000 × G for 15 min, and the precipitate was washed four times with distilled water. Microcrystalline cellulose, DCIP, and bovine serum albumin were purchased from Sigma-Aldrich Japan (Tokyo, Japan). Glucose and IPTG were purchased from Nacalai Tesque. NADH and NAD+ were purchased from Oriental Yeast Co. (Tokyo, Japan). Restriction enzymes were obtained from Takara Bio (Shiga, Japan). The other reagents were chemically pure grades of commercial products.
RESULTS AND DISCUSSION
Expression and purification of AZR-CBD.
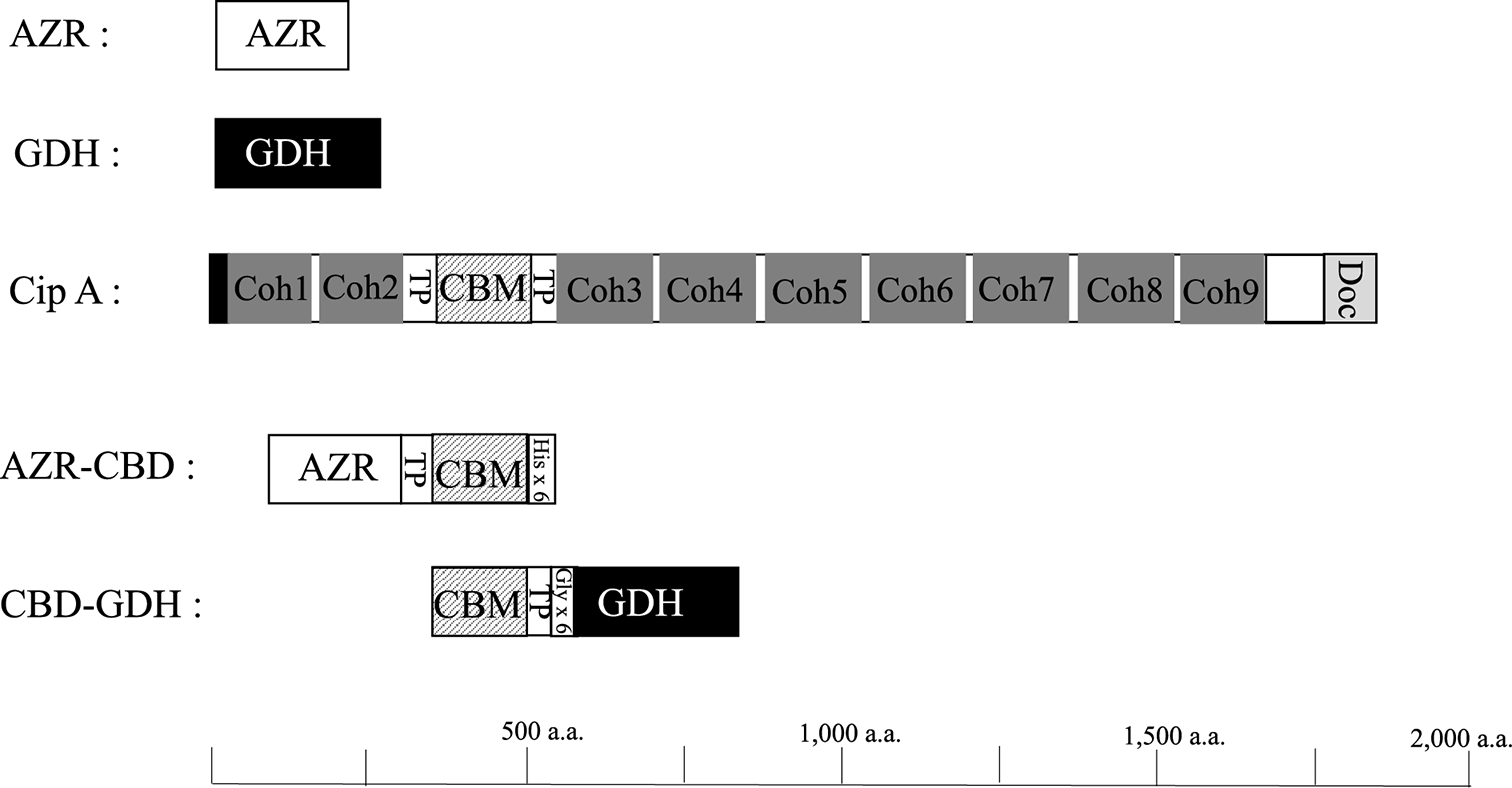
AZR from B. subtilis is a single-domain enzyme and consists of 211 amino acids. Cellulosome-integrating protein A (Cip A) of C. thermocellum is multi-domain enzyme and consists of two N-terminal cohesion domains, a first TP-rich linker, CBD classified CBM3, a second TP-rich linker, seven cohesion domains, and a C-terminal dockrin domain (Fig. 2). Based on the sequences of these proteins, an AZR and CBD fusion enzyme, AZR-CBD, was designed as N-terminal AZR, TP linker, CBD, and 6×His tag. To construct the fusion enzyme expression vector, initially, a gene with the first TP linker and CBD from Cip A were amplified by PCR and cloned into pET22b (+) vector (pET22b-cbdC). Subsequently, AZR gene of B. subtilis was cloned into pET22b-cbdC. The constructed plasmid, pET22b-azr-cbd, introduced into E. coli Rosetta-gami B (DE3), and significant AZR activity was detected in the cell-free extract (4.2 units/mg protein). AZR-CBD was purified using Ni-NTA column chromatography, and the specific activity of purified AZR-CBD was 15.1 units per mg of protein (Table S1; see J. Appl. Glycosci. Web site).
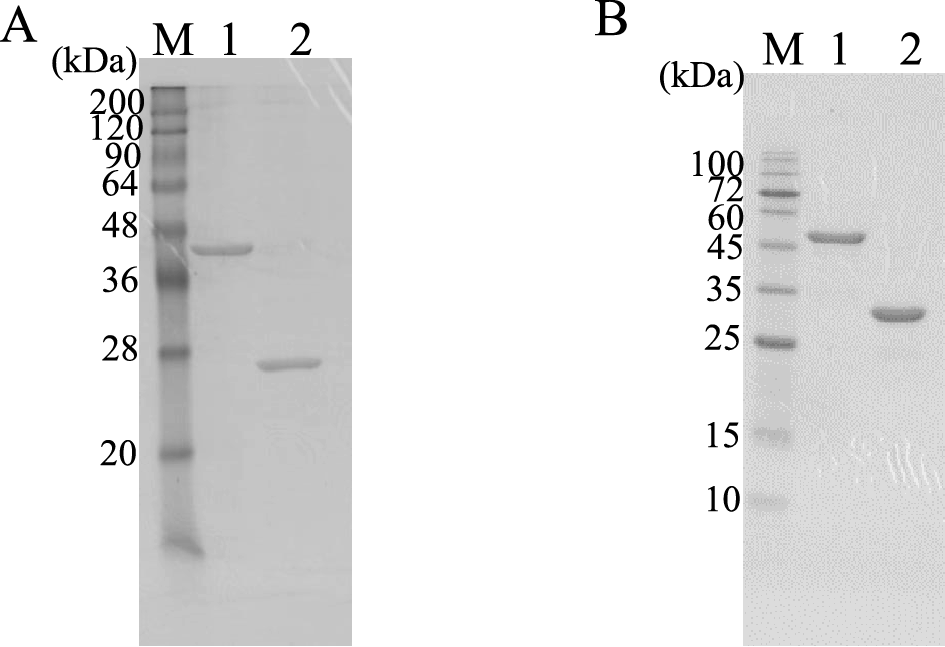
To compare AZR-CBD with wild-type AZR, AZR was also expressed in E. coli Rosetta-gami B (DE3) and purified from the cell-free extract using ion exchange and hydrophobic column chromatography (Table S2; see J. Appl. Glycosci. Web site). The purified AZR exhibited a specific activity of 65.3 units/mg. The predicted molecular masses of AZR-CBD and AZR based on their amino acid sequences were approximately 47 and 23 kDa, respectively. Their molecular masses estimated by SDS-PAGE were almost in agreement with these values (Fig. 3A). Although the size of AZR-CBD is two times larger than that of AZR, the specific activity of AZR-CBD was four times lower than that of AZR. This result suggested that the addition of CBD to AZR caused a decrease in specific activity. The additional CBD might prevent either substrate binding to the enzyme or product release from the enzyme. However, the activity of AZR-CBD was sufficient to perform the colorimetric determination of NADH.
Expression and purification of CBD-GDH.
The gene for GDH from B. subtilis consists of 783 bp nucleotides encoding 261 amino acids. In our preliminary experiments, we constructed GDH and CBD fusion enzymes consisting of N-terminal GDH, TP linker, and C-terminal CBD, but the fusion enzyme had no GDH activity. Therefore, the fusion enzyme (CBD-GDH) was constructed with N-terminal CBD, TP linker, Gly×6 repeats linker, and C-terminal GDH (Fig. 2). The CBD-GDH expression vector was constructed using the procedure described below. The CBD gene and second TP linker was ligated with pET 22b vector (pET22b-Ncbd). Subsequently, the GDH gene was amplified by PCR, and Gly×6 repeats linker were added upstream of the GDH gene by PCR. Finally, the PCR fragment was cloned into pET22b-Ncbd, and the constructed vector, pET22b-cbd-gdh, was introduced into E. coli Rosetta-gami B (DE3). To express wild-type GDH in E. coli, pET22b-gdh was also introduced into E. coli Rosetta-gami B (DE3). Both enzymes were purified from cell-free extract using ion exchange and hydrophobic column chromatography (Table S3 and S4; see J. Appl. Glycosci. Web site). The specific activity of CBD-GDH and GDH were 22.6 and 15.8 units per mg of protein, respectively. The predicted molecular masses of CBD-GDH and GDH based on their amino acid sequences were 50 and 28 kDa, respectively. As shown in Fig. 3B, the final preparations of CBD-GDH and GDH were homogeneous by SDS-PAGE, and their molecular masses agreed with the predicted molecular masses. The specific activity of CBD-GDH was higher than that of GDH. The addition of CBD might affect the stability of the enzyme. After hydrophobic column chromatography, the specific activity of GDH decreased, but that of CBD-GDH did not.
Cellulose binding activity of CBD-fusion enzymes.
Table 2 shows the binding activity of AZR-CBD to swollen cellulose and microcrystalline cellulose. AZR-CBD bound to swollen cellulose at a rate of approximately 97 %, but wild-type AZR hardly bound to it. This finding indicated that CBD gave the binding ability to AZR. Although AZR-CBD strongly bound to 0.5 % swollen cellulose, the binding efficiency of AZR-CBD to 0.5 % microcrystalline cellulose was only about 15 %. An increase in the amount of microcrystalline cellulose increased binding efficiency, and AZR-CBD bound to 10 % microcrystalline cellulose at a rate of approximately 44 %.
Table 2.
Cellulose binding activity of AZR and AZR-CBD
| Enzyme |
Substrate (final conc.) |
Binding
Efficiency
(%) |
| AZR |
Swollen cellulose (0.5 %) |
3 |
| AZR-CBD |
Swollen cellulose (0.5 %) |
97 |
| Microcrystalline cellulose (0.5 %) |
15 |
| Microcrystalline cellulose (2.5 %) |
18 |
| Microcrystalline cellulose (5 %) |
22 |
| Microcrystalline cellulose (7.5 %) |
37 |
| Microcrystalline cellulose (10 %) |
44 |
As shown in Table 3, the maximum amount of CBD-GDH also bound to swollen cellulose but GDH did not. For 0.5 % microcrystalline cellulose as a substrate, the binding efficiency of CBD-GDH declined to approximately 8 %. In the case of CBD-GDH, the binding efficiency was increased by the increase in microcrystalline cellulose, and about 99 % of CBD-GDH bound to 10 % microcrystalline cellulose.
Table 3.
Cellulose binding activity of GDH and CBD-GDH.
| Enzyme |
Substrate |
Binding
Efficiency
(%) |
| GDH |
Swollen cellulose (0.5 %) |
5 |
| CBD-GDH |
Swollen cellulose (0.5 %) |
98 |
| Microcrystalline cellulose (0.5 %) |
8 |
| Microcrystalline cellulose (2.5 %) |
27 |
| Microcrystalline cellulose (5 %) |
35 |
| Microcrystalline cellulose (7.5 %) |
62 |
| Microcrystalline cellulose (10 %) |
99 |
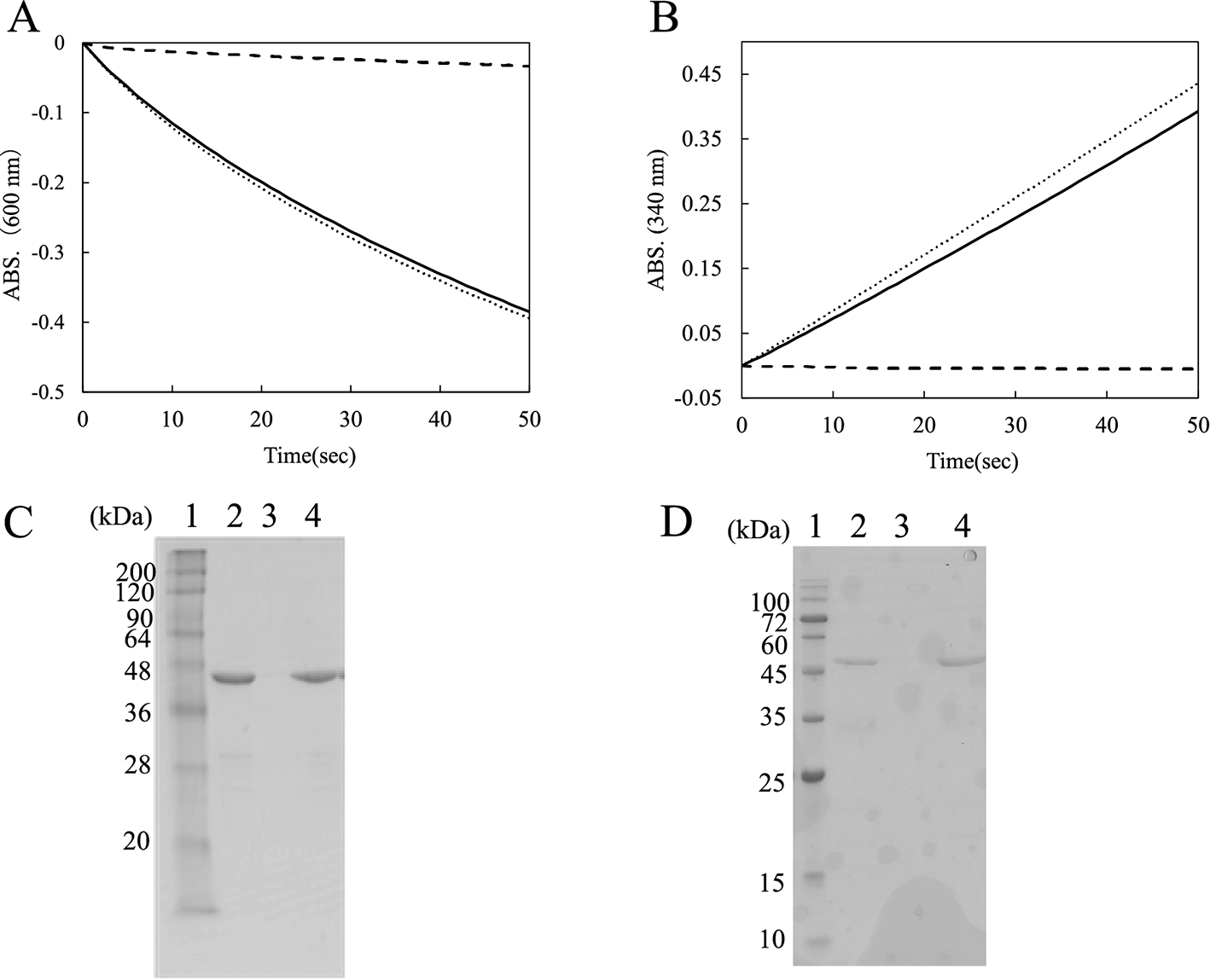
The enzyme activities of AZR-CBD and CBD-GDH were measured after immobilization on swollen cellulose. The mixtures of enzyme and swollen cellulose were centrifuged, and the precipitates were resuspended in buffer after centrifugation. Figure 4A shows the time course of reduction of DCIP with the supernatant and the resuspended precipitate. The supernatant scarcely showed any enzyme activity. On the other hand, the resuspended precipitates showed about 92 % reactivity compared with the control sample (without swollen cellulose). In the case of CBD-GDH, GDH activity in the resuspended precipitate retained about 88 % reactivity (Fig. 4B). Figures 4C and D show SDS-PAGE analysis of the supernatants and the resuspended precipitates. AZR-CBD and CBD-GDH were present in swollen cellulose in the resuspension but not in the supernatant. The findings suggested that both enzymes retained their own catalytic activity after binding to cellulose, and that the addition of CBD to these oxidoreductases allowed them to become immobilized on the cellulose without covalent binding or chemical cross-linking.
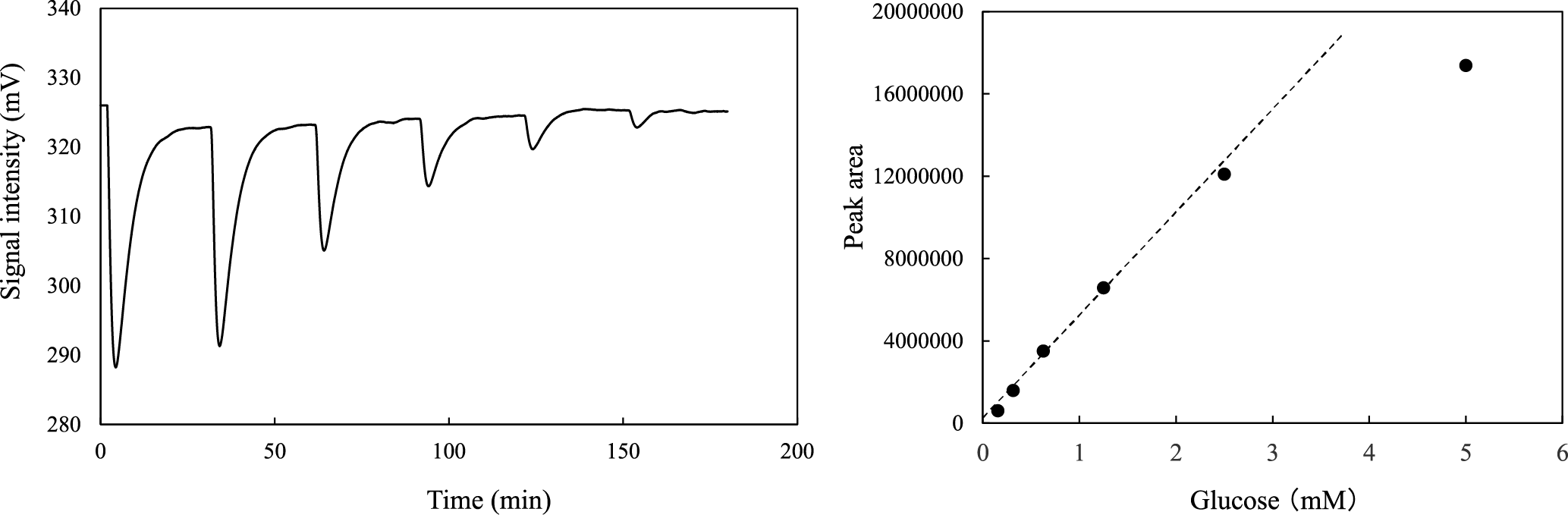
Flow injection analysis with AZR-CBD and CBD-GDH.
The decolonization of DCIP by reduction was caused by addition of AZR-CBD and CBD-GDH to the mixture containing glucose, NAD+, and oxidized DCIP (Fig.S1; see J. Appl. Glycosci. Web site). The finding suggests that glucose is detected by combining the AZR and GDH reactions. To prepare the reactor for flow injection analysis, AZR-CBD and CBD-GDH were immobilized on microcrystalline cellulose. In our preliminary experiments, swollen cellulose was used as the supports, but the outlet membrane filter of the reactor became clogged with swollen cellulose. Both enzymes were mixed with microcrystalline cellulose. The mixture of AZR-CBD and cellulose was poured into the reactor, and the mixture of CBD-GDH and cellulose was poured in subsequently. As a result, the reactor consisted of two layers; the top layer contained microcrystalline cellulose-immobilized CBD-GDH and the bottom comprised cellulose-immobilized AZR-CBD (Fig. 1B). As described above, AZR-CBD (0.56 mg/mL) and CBD-GDH (0.25 mg/mL) bound to 10 % microcrystalline cellulose at rates of 44 and 99 % of the initial protein amount, respectively. In the reactor, about 25 μg of AZR-CBD and CBD-GDH could be immobilized on the cellulose.
In our flow injection analysis system, a two-pump system was employed to suppress the background peak caused by water or buffer injection (Fig. 1C), and the total flow rate of the system was set at 0.5 mL/min (0.25 mL/min × two lines). The standard glucose solutions were injected into the deionized water line, and the solutions were mixed with buffer containing 80 μM DCIP oxidant and 1 mM NAD+. Figure 5A shows that a result of successive 50 μL injections of glucose solutions, and that the decrease in glucose concentration caused a decline in the intensity of the signal peak of DCIP reductant. This result indicated that glucose was oxidized to D-glucono-1,5-lactone and NAD+ was reduced to NADH by immobilized GDH in the top cellulose layer in the reactor. Subsequently, NADH produced by GDH was oxidized and DCIP oxidant was reduced by immobilized AZR in the bottom layer, and the decolorization of DCIP by reduction was monitored using a spectrophotometer. Figure 5B shows the relationship between the amount of glucose and the intensities of the signal peaks, and a linear calibration curve was observed in the range for about 0.16 to 2.5 mM glucose with a correlation coefficient, r, of 0.998. In contrast, the intensity at 5.0 mM glucose deviated substantially from the linear trace. In some cases of flow injection analysis with an immobilized enzyme, a lowering of sensitivity toward high substrate concentration was explained by the reaction kinetic of the immobilized enzyme (i.e., at high substrate concentration, the initial velocity was not sensitive to change in the substrate concentration). Our flow injection system was based on the coupled reaction of GDH and AZR, and the deviation in the intensity with 5.0 mM glucose from the linear calibration might be attributable to the kinetics of either GDH or AZR.
In conclusion, CBD fusion enzymes, AZR-CBD and CBD-GDH, were constructed, and the fusion enzymes showed cellulose binding activity. After immobilization on cellulose, CBD-GDH oxidized glucose and reduced NAD+, and also AZR-CBD oxidized NADH and reduced DCIP. Immobilized AZR-CBD and CBD-GDH could be applied for the determination of glucose concentration using a flow injection analysis system. The reactor for this system consisted of a two-layer structure, and the reaction product of the first-layer was readily catalyzed by the enzyme in the second-layer. These findings suggested that the CBD fusion enzyme was easily immobilized on cellulose, and this immobilization method permits the determination of multi-step enzymatic reactions. In this study, we selected the cellulose binding domain for addition of the binding ability to oxidoreductase. A large number of CBDs have been characterized, and some of them are available for the immobilization of enzymes. We are searching for combinations of CBD and polysaccharide that are suitable for enzyme immobilization. This approach may be useful for the construction of a reactor consisting of multiple layers of immobilized enzymes for the development of biosensors.
CONFLICTS OF INTEREST
The authors declare no conflict of interests.
ACKNOWLEDGMENTS
This work was carried out through collaboration of the Core to Core Program, supported by the Japan Society for the Promotion of Science and the National Research Council of Thailand.
REFERENCES
- 1) S.W. May: Applications of oxidoreductases. Curr. Opin. Biotechnol., 10, 370–375 (1999).
- 2) R. Shi, K. Stein, and G. Schwedt: Spectrophotometric determination of glucose in foods by flow injection analysis with an immobilized glucose oxidase reactor. Z. Lebensm. Unters. Forsch., 204, 99–102 (1997).
- 3) S. Arimori, C.J. Ward, and T.D. James: A D-glucose selective fluorescent assay. Tetrahedron Lett., 34, 303–305 (2002).
- 4) R. Nenkova, R. Atanasova, D. Ivanova, and T. Godjevargova: Flow injection analysis for amperometric detection of glucose with immobilized enzyme reactor. Biotechnol. Biotechnol. Equip., 15, 1986–1992 (2014).
- 5) S. Datta, LR. Christena, and Y.R.S. Rajaram: Enzyme immobilization: an overview on techniques and support materials. 3 Biotech, 3, 1–9 (2013).
- 6) A. Nakamura, T. Tasaki, D. Ishiwata, M. Yamamoto, Y. Okuni, A. Visootsat, M. Maximilien, H. Noji, T. Uchiyama, M. Samejima, K. Igarashi, and R. Iino: Single-molecule imaging analysis of binding, processive movement, and dissociation of cellobiohydrolase Trichoderma reesei Cel6A and its domains on crystalline cellulose. J. Biol. Chem., 291, 22404–22413 (2016).
- 7) M. Alahuhta, Q. Xu, Y.J. Bomble, R. Brunecky, W.S. Adney, S.Y. Ding, M.E. Himmel, and V.V. Lunin: The unique binding mode of cellulosomal CBM4 from Clostridium thermocellum cellobiohydrolase A. J. Mol. Biol., 402, 374–387 (2010).
- 8) J.E. Hyeon, S.D. Jeon, and S.O. Han: Cellulosome-based, Clostridium-derived multi-functional enzyme complexes for advanced biotechnology tool development: advances and applications. Biotechnol. Adv., 31, 939–944 (2013).
- 9) Y. Berdichevsky, R. Lamed, D. Frenkel, U. Gophna, E.A. Bayer, S. Yaron, Y. Shoham, and I. Benhar: Matrix-assisted refolding of single-chain Fv-cellulose binding domain fusion proteins. Protein. Expr. Purif., 17, 249–259 (1999).
- 10) Z. Gao, Q. Zhang, Y. Cao, P. Pan, F. Bai, and G. Bai: Preparation of novel magnetic cellulose microspheres via cellulose binding domain-streptavidin linkage and use for mRNA isolation from eukaryotic cells and tissues. J. Chromatogr. A, 1216, 7670–7676 (2009).
- 11) A. Kumar, S. Zhang, G. Wu, C.C. Wu, J. Chen, R. Baskaran, and Z. Liu: Cellulose binding domain assisted immobilization of lipase (GSlip–CBD) onto cellulosic nanogel: characterization and applicationin organic medium. Colloids Surf. B Biointerfaces, 136, 1042–1050 (2015).
- 12) A. Fishman, I. Levy, U. Cogan, and O. Shoseyov: Stabilization of horseradish peroxidase in aqueous-organic media by immobilization onto cellulose using a cellulose-binding-domain. J. Mol. Catal. B Enzym., 18, 121–131 (2002).
- 13) Y. Nishiya and Y. Yamamoto: Characterization of a NADH:dichloroindophenol oxidoreductase from Bacillus subtilis. Biosci. Biotechnol. Biochem., 71, 611–614 (2007).
- 14) R.F. Ramaley and N. Vasantha: Glycerol protection and purification of Bacillus subtilis glucose dehydrogenase. J. Biol. Chem., 258, 12558–12565 (1983).
- 15) O. Yaniv, E. Morag, I. Borovok, E.A. Bayer, R. Lamed, F. Frolow, and L.J.W. Shimond: Structure of a family 3a carbohydrate-binding module from the cellulosomal scaffoldin CipA of Clostridium thermocellum with flanking linkers: implications for cellulosome structure. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun., 69, 733–737 (2013).
- 16) J. Tormo, R. Lamed, A.J. Chirino, E. Morag, E.A. Bayer, Y. Shoham, and T.A. Steitz: Crystal structure of a bacterial family-III cellulose-binding domain: a general mechanism for attachment to cellulose. EMBO J., 15, 5739–5751 (1996).
- 17) O.H. Lowry, N.J. Rosebrought, A.L. Farr, and R.J. Randall: Protein measurement with the folin phenol reagent. J. Biol. Chem., 193, 265–275 (1951).
- 18) U.K. Laemmli: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685 (1970).
- 19) Y.H. Zhang, J. Cui, L.R. Lynd, and L.R. Kuang: A transition from cellulose swelling to cellulose dissolution by o-phosphoric acid: evidence from enzymatic hydrolysis and supramolecular structure. Biomacromolecules, 7, 644–648 (2006).
