We have applied alchemical free energy calculation of theophylline with an RNA aptamer with one or more ligand docking poses. We find that the predicted binding affinity strongly depends on the anchor's position at the receptor using only distance restraints; it means that a ligand is trapped at the other metastable states during the decoupling process. We also demonstrate that the binding affinity of a fragment-like molecule such as theophylline to the receptor is obtained by phase space decomposition with orientational restraints.
計算機システムの高速化,計算手法の進展によってFragment-Based Computer-Aided Ligand Designが現実のものになろうとしている.フラグメント分子は分子量が300程度と小さいため,結合親和性が小さく,かつ結合サイト内に複数の結合ポーズを持つことが予想される.そこで,我々は,対象としてテオフィリン分子/RNA複合体を用いて,フラグメントライクな分子に対するアルケミカル変換法による結合親和性(結合自由エネルギー)計算方法について調べた.
結合親和性の定量予測するために,アルケミカル変換法による結合自由エネルギー計算を行った.熱力学的サイクル中で標準状態との関係を維持するために,距離拘束ポテンシャルを用いた(Figure 1).ここで,標的分子側のアンカー原子群の位置を三種類変化させて自由エネルギー計算を行った(Table 1).

Thermodynamic cycle for alchemical free energy calculations.

Figure 1から標準結合自由エネルギーは,
テオフィリン分子/RNA複合体構造のモデル化には1O15 (PDB) [2]を用いた.RNAの力場はff14SBを使い,テオフィリン分子は点電荷にRESP,力場にGAFFを用いた.ユニットセル中に3個のMg2+イオンをGoudaら [3]のモデルに従って配置し,セルを中性化するように26個のNa+イオンを導入した.結合自由エネルギー評価のために構造サンプリングは,ストキャスティック動力学計算を用いて,時間刻み2 fsで100 fsごと構造をサンプリングした.最初の500 psのデータを捨て,6 nsまでのデータをMBARで解析した.なお,統計誤差を小さくするために,各原子の初期運動量を乱数で5種類発生させて用いた.計算に用いたアルケミカル径路は,結合パラメーターλ(≡(λR, λC, λLJ))で中間状態を記述し,(0, 0, 0) → (1, 0, 0) → (1, 1, 0) → (1, 1, 1) の径路に沿って,それぞれ不等間隔に11点,6点,15点とった.また,アンカー原子群の重心とテオフィリン分子の重心の距離を拘束するために導入した調和ポテンシャルのバネ定数は,その分散が熱揺らぎ程度になるように選んだ(Table 1).
各アンカー原子群に対して,得られた結合自由エネルギーとその成分をTable 2に示す.#1とそれ以外との自由エネルギー差が大きいことが分かる.

次に,我々はメタダイナミクス法を用いて結合ポーズ探索を行ったところ,結合サイト内に複数の結合ポーズを持つことが分かった.そこで,最も安定な自由エネルギーを与える結合ポーズP1と二番目に安定な自由エネルギーを与える結合ポーズP2について調べたところ,#1のアンカー原子群の重心からほぼ等距離に構造P1および構造P2の重心が存在していることが分かった.
これらのことから,距離のみを拘束した構造サンプリングでは,#1の場合,構造P2にトラップされる影響が大きく,他のアンカー原子群#2,#3では構造P2のポテンシャル谷の存在を認識できないため,その他の微細な自由エネルギーミニマムのトラップの影響が大きいことが示唆された.すなわち,#1の結合自由エネルギー推定値は,結合パラメーターλの変化とともに構造P1のポテンシャル谷から脱出したリガンド分子が準安定構造P2のポテンシャル谷に長時間トラップされた結果である(Figure 2,3参照).

Spherical shell about the center of mass of the anchoring atoms for structural sampling.
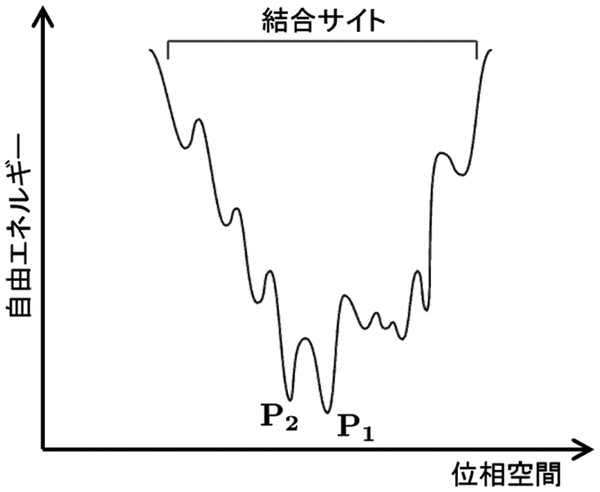
Schematic view of the binding free energy in #1.
一方,#1以外のアンカー原子群#2,#3の場合の結合自由エネルギー推定値は,構造P2のポテンシャル谷が認識されないため,それぞれのサンプリング空間(Figure 2参照)に存在する他の微小なポテンシャル谷にトラップされてしまうことを反映している.
この問題を克服するためには,
i. 結合サイトに対して,リガンド分子の相対的な"向き”を拘束して(P1あるいはP2の近傍の位相空間)計算を行うこと(位相空間分割法),
ii. 薄皮領域の全域を効率よく探索しながら,存在確率を評価することなどが考えられる.
ここでは,iの方法で計算を行った.リガンド分子の向きを調和ポテンシャルで拘束して,得られた自由エネルギー推定値は構造P1に対して−8.78 (0.04) kcal/mol,構造P2に対して−6.86 (0.07) kcal/molとなった.よって,系全体の結合自由エネルギーは−8.81 (0.04) kcal/molと見積もられる.
この結果は,実験値−8.9 kcal/molを非常に良く再現しており,フラグメントベースのドラッグデザインでは,距離のみの拘束では不十分で,向きを拘束して複数の結合ポーズで計算する位相空間分割法が有効であることが分かった.