近年急速に普及が進む電気自動車に対し,航続距離の長大化を実現できる電源として理論エネルギー密度が最も高いリチウム空気二次電池の開発が進められているが,十分な電流密度が得られておらず,電池性能の大幅な向上が求められている.そこで電池内の電流は電解質のイオンが担っていることから,水系リチウム空気電池の電解質に用いられるLiCl水溶液内のイオンの基礎的挙動について,分子動力学シミュレーションを行った.解析の結果,密度や自己拡散係数が実験値と定量的,定性的によい一致を示す結果が得られ,電解質の濃度が増加するほど水溶液中で多くのイオン対やイオンクラスターが形成されていることが分かった.
エネルギー環境問題の観点から,近年では電気自動車の開発と普及が積極的に進められており,現在はリチウムイオン二次電池が自動車用電源の主流として用いられている.しかし,リチウムイオン二次電池は大容量化と高速充放電の実現が依然として課題であるため,大幅な高容量化が実現できる次世代の電池として,リチウム空気電池が着目されている [1].リチウム空気電池はリチウム金属と空気中の酸素を放電反応に用いるため,単位重量・単位体積当たりの理論エネルギー密度が最も高い.しかし,現状のリチウム空気電池から得られる電流密度は理論値よりも大幅に低く,電池性能の向上が重要な課題となっている.
電池内の電流は電解質のイオンが担っていることから,リチウム空気電池の性能を決定づける因子として,電解質におけるイオンの挙動が挙げられる.特に水系電解質では溶質であるイオンと溶媒である水分子が静電相互作用などによって結びつき,水分子のネットワーク中にイオンが拡散している.この時,水分子は分極しているためにイオンが作る電場によって配向し,イオンを取り囲んで水和殻を形成する.その結果,イオンの振る舞いは水分子の影響を強く受け,さらに同種イオン間や異種イオン間の相互作用も合わさって複雑な挙動を呈する.したがって充放電時の電解質内のイオン輸送現象を検討するには,水系電解質内のイオンの基礎的挙動を分子スケールで理解することが必要不可欠である.
電解質中のイオンの挙動については,Debye-Hückel理論 [2]が最も有名であり,統計熱力学的な手法を用いて電解質の熱力学的量を解明している [3,4].この理論ではイオンはDebye 長で示唆される距離において反対符号のイオン雰囲気をまとい,その相互作用を受けると考えられる.しかしDebye-Hücke理論は希薄な強電解質を対象とし,溶媒を連続体と見なしているため,電解質の濃度が高い場合は破綻することが知られている.高濃度な水系電解質においては,分子スケールでのイオン間の相互作用やイオン-水分子間の相互作用の影響がより顕著に現れ,イオンクラスターの形成などの現象が起こることが実験や数値計算から示唆されている [5].これらの現象はイオンが分子スケールで他のイオンや水分子の影響を受けていることに起因しており,水系電解質を用いた電池内のイオンにおいても同様に分子スケールの影響を受けながら輸送されていると考えられる.
そこで本研究では,水系リチウム空気電池に用いられる電解質であるLiCl水溶液を対象とした分子動力学シミュレーションを行った.特に電解質の濃度をパラメータとして,Debye-Hückel 理論が適用できる希薄濃度から実際の電池に用いられる濃度まで変化させ,実験から得られているマクロな物性値と比較しながら,水系電解質内におけるイオンの基礎的挙動を明らかにすることを目的とする.
分子スケールでの数値計算手法として,一般的な分子動力学法の中でも解析時間を比較的長く取れる古典分子動力学法を用いた.本研究では河村らによって開発された古典分子動力学法プログラムであるMXDORTO [6]を用いてシミュレーションを行い,原子2個から構成される2体間ポテンシャルと,原子3個から構成される3体間ポテンシャル用いた [7].分子内,分子間の区別はせず,全原子に関する原子間相互作用のみを考慮し,分子内振動も考慮したフレキシブルなモデルとした.ポテンシャル関数の各パラメータは,分子構造や熱力学量等の実験値を再現するよう,経験的に決定した.
本研究では,式(1)に示す2体間ポテンシャルを用いた.
| (1) |
全粒子間に作用する2個の粒子間に働く相互作用を,式(1)の2体間ポテンシャル関数を用いて表している.ここで,第1項は静電(クーロン)相互作用,第2項はBMHポテンシャルによる近接反発項,第3項はvan der Waals力を表している.また,第4–6項はMorse型のポテンシャルにより,共有結合の動径部分を表す.i,jは粒子を表し,rijは原子間距離,f0は単位換算のための定数である.また,z,a,b,c,D,β,r3ijは,各原子間相互作用を表すパラメータであり,分子の性質を表す.z,a,b,cは全粒子に与え,D,β,r3ijは各共有結合に与える.本研究で用いた各パラメータの値をTable 1に示す.
| Two body term | H2O | Li+ | Cl− | |
| H | O | |||
| z | 0.46 | −0.92 | −0.18 | 0.09 |
| a [Å] | 0.035 | 1.728 | 1.900 | 0.035 |
| b [Å] | 0.044 | 0.1275 | 0.140 | 0.058 |
| c [(kcal/mol)1/2/Å−3] | 0.0 | 27.4 | 32.1 | 0.0 |
| Morse term | H–O | |||
| D1ij [kcal/mol] | 13711.0 | - | - | |
| D2ij [kcal/mol] | −523.0 | - | - | |
| D3ij [kcal/mol] | 8.30 | - | - | |
| β1ij [Å−1] | 7.40 | - | - | |
| β2ij [Å−1] | 3.13 | - | - | |
| β3ij [Å−1] | 12.80 | - | - | |
| r3ij [Å] | 1.283 | - | - | |
一方,H2O分子を扱うためには2体間相互作用のみでは不十分であり,結合の方向性を定めるために3体間ポテンシャルを導入する必要がある.本研究は式(2),(3)に示す3体間ポテンシャルを用いた.
| (2) |
| (3) |
ここでθijkは近接する粒子i,j,kの角度であり,3体間ポテンシャルにより共有結合の角度部分を表している.fk,θ0,gr,rmは,各原子間相互作用を表すパラメータであり,本研究で用いた各パラメータの値をTable 2に示す.
| Three body term | H–O-H |
| fk [10−19J] | 0.000115 |
| θ0 [°] | 99.50 |
| rm [Å] | 1.43 |
| gr [Å−1] | 9.20 |
計算領域は三次元周期境界条件とし,静電相互作用の計算はEwald法を,分子の運動は速度Verlet法をそれぞれ用い,粒子数N,圧力P,温度Tを一定としたNPTアンサンブルで計算した.圧力の制御は基本セルの形状スケーリングにて行い,温度の制御は粒子速度のスケーリングで行った.計算の時間刻みは0.40 fsとした.
2.2 解析モデルおよび解析条件本計算では水系リチウム空気電池の電解質に用いられるLiCl電解質水溶液を対象に,LiCl の濃度をパラメータとして解析を行った.詳細な解析条件をTable 3 に示す. 粒子数と温度,圧力を一定に保つ条件で解析を行い,粒子数は5000 個の水分子に対して1,50,100 個のLiCl を含む条件を設定した.多数の水分子を用いることでDebye-Hückel 理論が適用できる希薄濃度(0.01 M)条件を再現し,さらに実際に水系リチウム空気電池の電解液として用いられている濃度(0.5 M,1.0 M)にも対応させた [8].なお温度と圧力は,室温および大気圧条件とした.
| Case | NH2O | NLi+ | NCl- | C [M] | T [K] | P [MPa] | dt [fs] | t [ps] |
| 1 | 5000 | 1 | 1 | 0.01 | 300 | 0.1 | 0.4 | 400 |
| 2 | 5000 | 50 | 50 | 0.5 | 300 | 0.1 | 0.4 | 400 |
| 3 | 5000 | 100 | 100 | 1.0 | 300 | 0.1 | 0.4 | 400 |
解析に用いる初期構造データの作成手順として,Table 3の各条件における予備計算によって安定するVを求めた後,T = 400 Kの条件でNVT アンサンブルを用いて粒子位置のランダマイズを行った.次に再びTable 3の条件においてNTPアンサンブルを行い,平衡状態に達した結果を初期構造データとした.初期構造データから計算を400 ps間行った結果を対象として解析を行った.実際に計算を行った濃度1.0 Mの解析モデルをFigure 1 に示す.Cl−およびLi+が計算領域全体に分散している様子が見られる.なお,Table 3の各粒子数の条件に対して,それぞれ初期構造データを5 セット作成し,200 step 毎の位置データを用いて統計量を取得した.

Snapshot of LiCl electrolyte (O:yellow, H:blue, Cl−:green, Li+:red).
計算の結果得られた各イオン種,各濃度における2 体間相関関数(PCF)から,LiCl水溶液内の水分子とイオンの分布について考察する.Figure 2 はH2OについてのPCF を示しており,特にFigure 2 (b)ではO–H 結合を示す大きなピーク(r = 0.93 Å)と,水分子間の構造化を示すピーク(r = 1.85 Å,3.22 Å)が見られ,液体状態の水分子間の構造が再現されている.さらにPCFは濃度を変えても同じ分布を呈しており,本計算条件で用いた濃度範囲では水分子同士の構造は変化していないと考えられる.

Pair correlation function of (a) O–O, (b) O–H, (c) H–H.
Figure 3 はCl−とLi+ に関するPCF である.Cl−とLi+は強電解質に分類されるため,水溶液中で完全に電離していればPCF = 1になるはずであるが,0.5 Mおよび1.0 Mの条件でCl−–Cl− 間,Li+–Li+ 間,Cl−–Li+ 間の全てにおいて第1 ピークが生じている.特にCl−–Li+ 間のPCFのピーク値が大きく,Cl−とLi+のイオン対が形成されていると考えられ,さらに第2,第3 ピークの存在から,イオン対よりも大きなイオンクラスターの形成も示唆されている.

Pair correlation function of (a) Cl−–Cl−, (b) Li+–Li+, (c) Cl−–Li+.
またFigure 3において濃度に対するPCFの第1ピーク値の変化に着目すると,Cl−–Cl− 間やLi+–Li+ 間は1.0 M,Cl−–Li+間では0.5 Mで最大値を取っており,異なる傾向を示している.しかしPCFの定義は,計算領域内のイオンの数密度に対する距離Rでのイオン分布の偏りを表しているため,濃度条件が異なる場合はイオン間のPCFピーク値を注意深く比較しなければならない.
例えば濃度条件が変化してもPCFのピーク値が同じだった場合は,あるイオン周りにおける他のイオンの存在確率は濃度に比例していることを意味する.Cl−–Cl− 間とLi+–Li+ 間のPCFは濃度が高いほど第1ピーク値が増加していることから,単純にイオン同士が近接する確率が増えるだけでなく,イオンクラスターも形成され,イオン分布の偏在性が強くなっていることを示唆している.
一方,Cl−–Li+間のPCFは1.0 Mの方が0.5 Mの場合より第1ピーク値は小さい.これは,Cl−周りでのLi+の存在確率が0.5 Mと1.0 Mであまり差異が無いことを表している.もしLi+の存在確率に濃度依存性が全くなければ,0.5 Mに対して1.0 Mは濃度が2倍であるため,PCFのピーク値は1/2倍になるはずであるが,実際には1.0 Mのピーク値は0.5 Mのピーク値の半分よりもやや大きい.したがって,高濃度のLiCl水溶液ほどCl−Li結合が形成されやすくなっていると考えられる.
水溶液中の電離状態に関する報告によれば,希薄な極限状態ではアニオンとカチオンはそれぞれ独立に水和されているが,アニオンとカチオンが直接あるいは水分子を一つ挟んでイオン対を形成している状態も準安定であり,さらに有限の濃度ではイオン対を作っているもののほうが多くなる [9,10].完全電離を仮定しているDebye-Hückel の理論値が0.01 M を超える濃度では実測値と大きくずれることも,本計算で示されるようなイオン対やイオンクラスターの形成に起因すると考えられる.
Figure 4とFigure 5はそれぞれCl− とH2O間,Li+ とH2O間のPCF である.いずれもCl− とLi+周りに水和殻形成を示唆する第1,第2ピークが存在していることが分かる.濃度が増加すると各PCFの第1ピークの値は減少する一方,特にCl−–O 間とCl−-H 間では新たなピークが出現するなど,顕著な変化が見られる.Figure 3において述べたように,濃度の増加に伴ってCl−とLi+のイオン対が形成される結果,Cl−とLi+に水和する水分子の数が減少するため,それぞれのPCFのピーク値が減少したと考えられる.
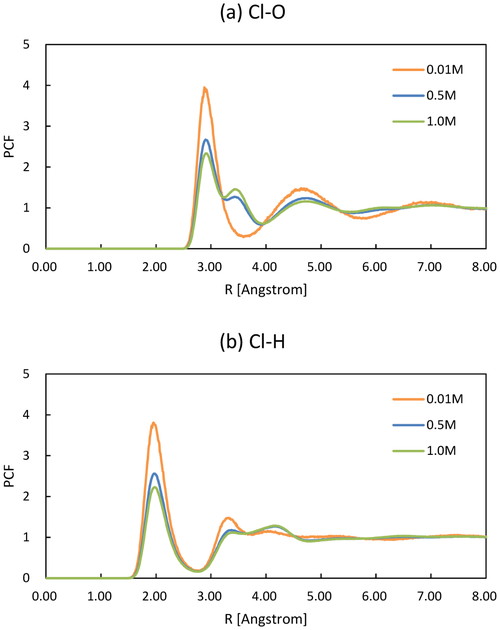
Pair correlation function of (a) Cl−–O, (b) Cl−–H.

Pair correlation function of (a) Li+–O, (b) Li+–H.
Figure 2からFigure 5まで示したPCFの第1 極大までの距離(rmax)をTable 4 に示す.これまでに述べたように,O–Cl−とO–Li+ のrmaxはイオンの水和状態を表していると考えられる.そこで各イオンの結晶半径(rc)と水分子の半径(1.40 Å)の和(rA−OH2) [3]をイオン-水分子間の理論距離として,計算結果と比較する.O–Cl− の場合はrmax が2.88∼2.92 Åであるのに対してrA−OH2 は3.14 Åである一方,O–Li+ のrmax は2.06∼2.08 Åで,rA−OH2 は2.18 Åである [3].したがって,いずれもイオンと水分子との距離は理論値よりも若干近づいていることが分かる.
| Concentration[M] | |||
| 0.01 | 0.5 | 1.0 | |
| O (H2O)–O (H2O) | |||
| rmax[Å] | 2.77 | 2.77 | 2.77 |
| rmin[Å] | 3.37 | 3.37 | 3.38 |
| NC | 4.62 | 4.58 | 4.58 |
| Cl−–O (H2O) | |||
| rmax[Å] | 2.88 | 2.90 | 2.92 |
| rmin[Å] | 3.60 | 3.29 | 3.23 |
| NC | 6.29 | 4.19 | 4.41 |
| Li+–O (H2O) | |||
| rmax[Å] | 2.06 | 2.06 | 2.08 |
| rmin[Å] | 2.92 | 2.88 | 2.88 |
| NC | 4.66 | 3.28 | 2.88 |
| Cl−–Li+ | |||
| rmax[Å] | - | 2.02 | 2.04 |
| rmin[Å] | - | 3.10 | 3.11 |
| NC | 0.00 | 0.857 | 1.17 |
現在良く知られている水和のモデルとして,Franc-Wen 及び,Samoilov の水和モデルがある [11,12].これらのモデルではイオン半径と価数によって水和の特性が決まるが,近年の報告 [13]によれば,イオン半径と価数だけでなく電子状態も水和特性に影響を与えている可能性が示唆されている.したがって,イオンの水和によって水分子の電子状態に違いが生じ,Li+ では半径5 Å程度の範囲内の水分子まではイオンの電荷の一部を受け持つため,イオンの価数とイオンが実質的に持つ電荷は完全には一致しない可能性があり,今回の計算で用いたモデルについても静電相互作用が強い可能性がある.
Table 4には各濃度の計算結果から得られた原子やイオン種ごとの配位数(NC)も示している.これは各粒子種の積算配位数(RCN)を求めた後,Figure 2からFigure 5までの第一極小を示す距離(rmin)の値から求めた.
O (H2O)–O (H2O) のNC から,水分子は4∼5 の他の水分子に囲まれていることが分かる.氷は水分子が正四面体構造をとることで形成されるが,液体の水分子はその構造が壊れて存在しているため,4以上の値になったと考えられる.
Cl−–O (H2O)とLi+–O (H2O) のNCはそれぞれCl−とLi+の水和数を示している.0.01 MではCl−のNCが6∼7,Li+のNCが4∼5の間で推移しており,既報の結果 [3]と同等の値を示している.一方,0.5 Mと1.0 MではCl−とLi+はいずれも0.01 Mの場合よりもNCが低下している.これは電解質の濃度増加に伴って形成されるイオン対も増加した結果,イオンに近接できる水分子の数が減少したことが考えられる.
Cl−–Li+とLi+–Cl− のNC からは,あるイオン一つに対する対イオンの平均結合数が分かる.0.01 Mの場合はCl−–Li+,Li+–Cl−のNCは存在しないが,濃度の増加に伴ってNC は増加しており,前述したようなイオン対の形成が直接的に確認できる.
3.3 イオン対とイオンクラスターの形成これまでに示した結果から,LiCl水溶液では濃度の増加に伴って多くのイオン対やイオンクラスターが形成されることが分かった.そこで形成されているイオンクラスターの大きさやその存在比率を評価するため,一つのイオンが結合している対イオン数の割合を調べた結果をFigure 6に示す.濃度の増加に伴い,結合していない孤立したイオン,および一つの対イオンとのみ結合しているイオンの割合が減少していることが分かる.その一方で二つ以上の対イオンと結合しているイオンの割合が増加していることが分かる.したがって,濃度が増加するにつれて単純に対イオンが形成されるのではなく,イオン対同士の結合数が増え,イオンクラスターが形成されていくと考えられる.

Number of Li–Cl bondings around (a) Cl− and (b) Li+.
各濃度における密度の計算結果,および文献値 [14]をFigure 7 に示す.密度は0.01 Mから1.0 Mに至るまで定性的,定量的に極めて良く一致していることが分かる.また,Figure 2において水分子間の構造に濃度依存性が見られないことから,Cl−とLi+は水分子同士の隙間に入り込むように分散していると考えられる.

Density of Li–Cl electrolyte vs. concentration.
Figure 8は各濃度における自己拡散係数を示しており,計算結果から得られた平均二乗変位の勾配を基にEinsteinの式を用いて算出した.計算から得られたそれぞれの自己拡散係数は,いずれも実験値 [15]より小さい傾向が見られる.これは本計算で用いた水のポテンシャルモデル [16]において,自己拡散係数がやや小さく見積もられていることに起因すると考えられる.また3.2で述べたように,Cl−およびLi+に対する水分子の距離(rmax)が理論値(rA−OH2)よりも小さく,各イオンが水分子を引きつける力が若干強いことが示唆されているため,より遠方の水分子も引きつけられ,第一水和圏だけでなく第二,第三水和圏も形成された結果,イオンの移動度が低くなったと推定される.またこれまで述べてきたように,LiCl水溶液中ではCl−–Li+結合が多数存在していることが示唆されており,イオン対やクラスターが形成されることで単独のイオンよりも水中での移動度が低下し,自己拡散係数も小さくなったと推定される.なお今回比較対象とした実験値 [15]を含め,LiCl水溶液を対象とした自己拡散係数の精緻な計測実験例が非常に少なく,実験値そのものの精査も今後必要であると考えられる.

Diffusion coefficients vs. concentration.
一方,本計算で用いた水のポテンシャルモデルは誘電率の再現性が極めて高いことが示されている [16].したがって,誘電率によって決定づけられる水分子間の構造やイオンの水和状態については,妥当な結果が得られていると考えられる.これは水分子およびCl− の自己拡散係数が減少する傾向が定性的に良く一致していることからも,濃度の増加に伴って水和殻半径が増大する様子が適切に再現されていると考えられる.
水系リチウム空気二次電池の電解質に用いられるLiCl水溶液内のイオンの基礎的挙動について,分子動力学シミュレーションを行った.解析の結果,電解質濃度によらず水分子間の構造がほとんど変化していない一方,強電解質であるCl−とLi+がイオン対やイオンクラスターを形成しながら存在していることが分かった.さらにバルクの物性値として実験的に計測できる密度や自己拡散係数と計算結果を比較した結果,定量的,定性的によい一致を示す結果が得られた.
本研究の一部は科学研究費補助金(基盤(A) 15H02347)の助成を受けて実施した.ここに記して謝意を表する.