研究論文
反復構造を持つ高分子に対する分子動力学計算のためのパラメータ設定支援プログラムo2pの開発
2017 年 16 巻 3 号 p. 63-69

詳細
2017 年 16 巻 3 号 p. 63-69
Molecular dynamics (MD)の実行に必須の結合角や電荷などのパラメータセット(力場)がライブラリに存在しない場合,力場の量子力学計算による算出やパラメータ自動生成ツールの活用という手法が用いられる.しかし,これらの手法は原子数の多い高分子の場合には適していない.そこで,3つの単位構造(両末端の単位と中央部の繰り返し単位)を含むオリゴマーの力場を算出し,それらをポリマー形式(両末端部+中央部 × n)に変換し対処することがよく行われる.しかしながら,変換は手作業にならざるを得ず,非効率的でヒューマンエラーの恐れもある.そこで,反復構造を持つ高分子に対するMDを簡便かつ確実に実行すべく,分子動力学ソフトとして汎用されているGromacs用の力場変換半自動化プログラムo2pを開発した.開発にあたっては,作業負荷の低減と時間短縮を意図してGUIを積極的に取り入れた.また,複雑なファイル変換過程を自動化することによってヒューマンエラーの一掃を目指した.本プログラムにより変換したファイルを用いてノナン中のアミロース分子を想定した系でMDを実行したところ,実験による報告と同じく左巻きの一重らせん構造が形成され,プログラムが有効に動作していることを確認した.
MD計算によるダイナミクスの解析はタンパク質や核酸に対して頻繁に行われており,各種アミノ酸およびヌクレオチドに対する力場パラメータのライブラリは充実している.一方,合成高分子については,繰り返し単位の種類や重合度が多様である.また,多糖についても主要な単糖のライブラリは存在しているものの,単糖の種類と立体配座および結合様式は極めて多様でライブラリでは到底網羅できない.ライブラリにパラメータが存在しない場合には,一般的には①既存のパラメータライブラリから類似の部分構造をもつものを複数抽出してパラメータを合成,②量子力学計算で算出,③パラメータ自動生成ツールの活用,という方法のいずれかが用いられる.①の方法は,非常に手間と労力を要し,人為的ミスの懸念を脱しきれないため,我々は②や③の方法に着目した.しかし②の方法では,特に高分子の場合,計算コスト過多による算出不能に陥ってしまう.③の方法では,Antechamber [1]やSwissParam [2]などのパラメータ自動生成プログラムがしばしば用いられるが,これらはいずれも低分子向けのツールであり,高分子のパラメータの取得は困難である.そのため,3つの単位構造(両末端の単位と中央部の繰り返し単位)からなるオリゴマーを定義し,パラメータを算出した後に,オリゴマーのパラメータファイルを基にポリマー形式(両末端部+中央部 × n)のファイルを構築するという方法が考えられる.分子動力学ソフトとして汎用されているGromacs [3]で③の方法を利用した場合の一般的なポリマーパラメータの構築手順をFigure 1に示す.ここではこれら手順の詳細を細かく述べることはしないが,多くのファイル生成や作業が必要である.手順中の主な工程は3つである.一つ目は,オリゴマーのPDBファイルなどの構造ファイル中の各原子を各末端構造および繰り返し構造のいずれかとして定義する工程である(①Definition of repeating structure).二つ目は,オリゴマー中の繰り返し構造を反復して,ポリマーの構造ファイルを構築する工程である(②Creation of polymer structure file).この工程を広く用いられているAvogadro [4]などのモデリングソフトウェアで行うと,構造ファイル中の繰り返し構造の情報,例えば残基名などがGromacsで計算可能なポリマーのフォーマットに対応しない.三つ目は,オリゴマー全体について生成されたパラメータファイルをGromacsが一つ目で定義した部分構造として利用できるようポリマーのフォーマットへ変換する工程である(③Files conversion).ここでは,複数のファイルを作成する工程があり,そのために膨大な文字置換と重複の検出およびその排除を行なう必要が生じる.従来はこれらの煩雑な作業を手作業でせざるを得ず,人為的なミスを生じる原因となり高分子への適用は容易ではない.

Procedure to prepare a set of files required for MD simulation of a polymer from files of corresponding oligomer.
この困難を克服して高分子へのMDシミュレーションの適用性を飛躍的に高めるべく,Gromacsに適合した構造のパラメータ変換(オリゴマー形式 → 直鎖状ポリマー形式)プログラム(o2p)の開発を目指すこととした.開発にあたっては,①Definition of repeating structureにおいてはオリゴマー中の各原子の各構造単位(両末端,繰り返し構造)への帰属にGUIを導入して直感的な操作を具現化,②についてはポリマーの構造ファイル構築の自動化,③についてはパラメータ変換作業の自動化,を行った.
本プログラムはウェブアプリケーションであり,PHP [5],HTMLおよびJavaScriptを用いて作成した.グラフィカルについてはJavaScriptの3DライブラリであるThree.js [6]を用いた.パラメータファイル,PDBファイルおよび原子の帰属情報(末端構造あるいは中央構造を成す繰り返し単位のいずれに属するかに関する情報)は,ウェブサーバ上で動作するデータベース(PostgreSQL) [7]で管理した.ユーザは,AntechamberやSwissParamなどのプログラムにより作成したオリゴマーのファイルを,本プログラムに登録すれば,処理を経て登録した分子に関わる一連のMD計算用ファイルをダウンロードによって取得することが出来る.
本プログラムは先述の①~③に対応した下記の(1)~(3)の主要機能と補助機能(4)を有する.
(1) 繰り返し構造セレクタ
(2) ポリマージェネレータ
(3) ファイルコンバータ
(4) 溶媒・イオンアペンダ
プログラム中でのデータの主な流れをFigure 2に示す.

Major flow of data in the program.
ユーザはstep1で最低3単位を含むオリゴマーのパラメータファイル(Gromacsフォーマット)とそれに対応する構造ファイル(現在はPDBフォーマットのみ対応)をアップロードする.これらのファイルはユーザが作成することも出来るが,AntechamberやSwissParamなどのプログラムを用いて取得すれば良い.次にstep2で繰り返し構造セレクタを用いて,オリゴマーの全原子を末端構造および中央部の繰り返し構造に帰属する.そしてstep3で中央部の繰り返しの回数や,必要に応じて溶媒やイオンを指定すると,ポリマージェネレータ,ファイルコンバータ, 溶媒・イオンアペンダが自動的にポリマーのPDBファイルとGromacsの計算に必要な一連のファイルを生成する.ユーザはこれらのファイルをダウンロードして計算を実行できる.
各機能の詳細を以下に記す.
3.1 繰り返し構造セレクタ繰り返し構造セレクタはオリゴマーを3つの単位構造に特定する作業を容易にするためのグラフィカルなプログラムである.オリゴマー形式からポリマー形式へと変換したPDBファイルと共に一連のGromacs用計算ファイルを作成するためには,オリゴマーのPDBファイル中の全原子を末端構造あるいは繰り返し構造に正しく帰属させなければならない.オリゴマーのPDB ファイルは原子と座標が列挙されているテキストファイルであり,テキストファイル中に書かれた各原子がオリゴマーのどの部位にあるかを特定するにはモデリングソフトで表示した分子構造を見ながら各原子との照合を行う必要がある.また,ポリマー形式に対応したGromacsのパラメータの作成にあたり,どの繰り返し構造のどの部位を構成する原子であるか識別できるよう各々の原子および単位構造に固有のIDを付与しなければならない.そこで,オリゴマーの3次元構造を表示し,繰り返したい構造の両末端にある2原子をクリックすれば半自動で各原子を繰り返し構造と末端構造に帰属するとともに各構造単位を構成する各原子に固有のIDを自動発行する機能を持つ「繰り返し構造セレクタ」を開発した.Webブラウザから操作する際の実際の画面イメージをFigure 3に示す.
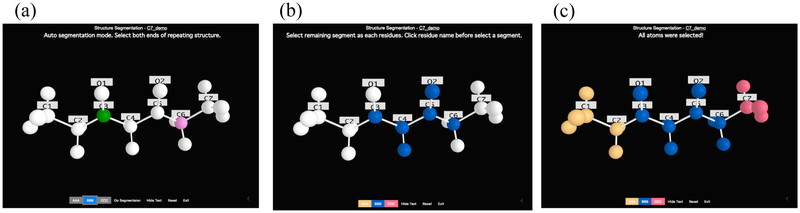
GUI for defining the repeating units and atoms.
ユーザがオリゴマー中の"中央部の繰り返し単位”にあたる構造の両末端にある2原子を指定してクリックすると,選択された原子は緑色と桃色で表示される(Figure 3 (a)).すると,クリックされた2つの原子とその間の原子が"中央部の繰り返し単位”を構成する原子として判定・定義され,青色で表示される.プログラムが繰り返し単位として判定できなかった原子は白色で表示される(Figure 3 (b)).これらの未定義の原子群はFigure 3 (b) O1のように繰り返し構造に含まれる場合と,C2などのように末端構造に含まれる場合がある.ユーザは未定義の原子が末端構造あるいは繰り返し構造に属するかを決定する必要があるが,その構造中の1つの原子を選択すれば,プログラムは原子結合を辿り未定義の原子群を末端構造あるいは繰り返し構造として定義する(Figure 3 (c)の黄色,赤色の表示原子).各原子に対するIDの自動設定は分子中の全原子が末端構造か繰り返し構造のいずれかに帰属されたことを自動判定した後に実行される.この繰り返し構造セレクタにより,これまで手動によりテキストエディタとモデリングソフトを見比べながら行っていた煩雑な作業を行うことなく,直感的に構造単位およびその構成原子の識別を定義することが可能となった.
3.2 ポリマージェネレータ「ポリマージェネレータ」は繰り返し構造セレクタで得た末端構造と繰り返し構造の帰属情報を用いて,ポリマー用のPDBファイルを作成するプログラムである.「繰り返し構造セレクタ」機能によって定義した繰り返し構造の空間座標を重合度に応じた回数平行移動させ,ポリマー構成原子の3次元座標を作成するとともに,ポリマー対応のパラメータに準じたフォーマットでPDBファイルを作成する.PDBファイルのみアップロードした場合でも,繰り返し構造を反復する回数(重合度)を任意に設定してポリマーのPDBファイルを得られる.
3.3 ファイルコンバータオリゴマーのパラメータファイルはポリマーのフォーマットとは異なっているため,そのままではポリマーのシミュレーションを実行できない.そこで,オリゴマーのパラメータファイルの形式をポリマー用のファイルに変換する必要がある.ファイルコンバータはユーザがアップロードしたオリゴマーのパラメータファイルのデータを基にポリマー用のファイル一式を自動的に作成するプログラムである.ポリマーに対応した複数のファイルにはそれぞれ異なった種類のパラメータが記されており,単純な構成のオリゴマーのファイルをそのまま変換できるわけではない.さらに,オリゴマーのパラメータでは各原子を通し番号で区別しているのに対し,Gromacsのポリマー対応パラメータファイルではatomtypesで区別する仕様であるため,通し番号からatomtypesへの置換も必要である.手動での置換の際にはパラメータの重複が不可避で,その検出と削除も行わなければならない.加えて,繰り返し構造と末端構造をGromacsに識別させるために,各構造に対してIDを付与するとともにatomtypesとの対応や各構造の連結部の原子の情報等を記したファイルも構造ごとに作成しなければならない.一連の煩雑な作業を担うのが「ファイルコンバータ」機能であり,ユーザがアップロードしたオリゴマーのパラメータファイルのデータに基づいてポリマー用のファイル一式を自動的に作成する.
3.4 溶媒・イオンアペンダ生体高分子のMD計算は溶媒を含む系で実行する場合がほとんどで,真空中でのシミュレーションはあまり行われない.溶媒分子を含む系での計算では溶媒分子に対するパラメータが必要である.Gromacsで溶媒分子を認識させるためには指定のフォーマットに則した複数のファイルを作成しなければならない.その作成を支援するのが「溶媒・イオンアペンダ」という補助機能である.ユーザがあらかじめ求めた種々の溶媒分子のパラメータを登録しておけば,必要に応じて溶媒を選択するだけでファイルが作成される.溶媒に限らず系にイオンを添加する際も,同様の手順で対応することができる.
開発したプログラムを用い,重合度(26 mer,38 mer,50 mer)の異なるアミロースについてノナン中でのMDシミュレーションを試みた.アミロースは非極性溶媒中やイオン溶液中で二重らせんであるA,B型ないし一重らせんのV型の立体構造を形成することが知られている [8].
MDシミュレーションの手順を以下に記す.
4.1 パラメータの登録糖などの立体構造ファイルや力場を提供する代表的なウェブアプリケーションの1つであるGLYCAM [9]からアミロース 8 merの構造ファイルを取得し,パラメータ決定プログラムSwissParamを用いてパラメータファイルを得た.1–4結合したα-グルコースは6残基で一回転するらせん構造をとることから,6残基を繰り返し構造とし,両端に1残基ずつのα-グルコースを付加した8残基でパラメータを算出した.SwissParamの出力ファイルの中からPDBファイルとGromacs用のパラメータが記載されたitpファイルを本プログラムにアップロードし,分子の登録を行った.ノナンのパラメータファイルもSwissParamで取得し,溶媒として登録した.
4.2 繰り返し構造の指定分子の登録を行った後,繰り返し構造セレクタのGUIを用いて内部構造を指定した.すなわち,繰り返し構造(中央の6 糖)の両端の原子(還元末端側にある1位のOと非還元末端側にある4位のC)の像をモニター上でクリックすることで,繰り返し構造を定義した.また,繰り返し構造に含まれない両末端の領域(残基)については,還元末端残基および非還元末端残基として定義した.
4.3 計算実行用ファイルの取得繰り返し構造の反復回数(重合度)を指定するとともに,溶媒としてノナンを選択し,PDBファイルとポリマー様式に整えられたフォーマットのパラメータをダウンロードした.今回の場合,ポリマーの重合度は26 mer,38 merおよび50 merであり,反復回数はそれぞれ4,6,8である.
4.4 MD計算の実行得られたファイルを用い,Gromacs-5.1.2で各モデルのMD計算(40 ns)を実行した.計算条件はLópezらの論文 [10]を参考に設定した.ただし,本計算では計算負荷の削減のため,分子から周期境界条件の壁面までの距離を2 nmから1 nmに変更した.既存の分子モデルビューワであるPyMOL [11]でシミュレーションによる立体構造の推移を調べた(Figure 4).いずれのモデルも分子内部で水素結合が形成されることで [8,12]分子の末端部を始点に徐々に初発の緩やかならせん構造から密ならせん構造への遷移が始まり,22 ns以内に安定な左巻きの一重らせん(V型)構造に達した.最も重合度の高いモデルである50 merにおけるらせん構造の1巻きの長さは8.0Å と算出され(Figure 5),X線構造回折の解析から得られている長さである7.91–8.17Å [12]とほぼ一致した.これら一連の操作により,開発したソフトウェアを用いたMDシミュレーションが的確に行われていることが立証された.

Snapshots of amylose molecules with different lengths during the MD simulation (in nonane).

Structure of amylose (50 mer) in nonane.
本プログラムにより,オリゴマーのパラメータとPDBファイルから対応する重合度の異なるポリマーのMD計算に必要なファイルを容易に作成することが可能となり,MD計算を実行するまでの過程をより迅速に行えるようになった. ただし,本プログラムを運用するうえで留意しなければならない点が二つある.一つはMD計算における重要なパラメータである電荷の扱いである.シミュレーションを実行するには系全体の電荷が0でなければならず,電荷を有するモデルに対しては,電荷を相殺するためのカチオンやアニオンの系への添加が必須である.したがって,モデルの総電荷は常に整数でなくてはならない.ところが,オリゴマーのパラメータを本プログラムでポリマー様式に変換する場合,必ずしも繰り返し構造の電荷が整数となるとは限らず,したがってシミュレーションの対象である高分子モデルの電荷も必ずしも整数とはならない.指定した構造(末端構造および繰り返し構造)ごとの電荷はプログラムによって表示されるものの,それが適切か否かの判定および不適切な場合の修正はプログラムでは行うことができない.したがって,適切な電荷を導けるよう事前に繰り返し構造の電荷を整える工夫が場合によっては必要である.もう一つの留意点は生成されたPDBファイルに基づくポリマーの立体構造である.ポリマー用PDBファイルはもととなるオリゴマーモデル中の繰り返し構造の単純な平行移動連結によって作成する.繰り返し構造が側鎖を有する場合,単純な平行移動と連結では側鎖の接触が起きることがある.分子構造の破綻の検出や修正を行う機能は備わっていないので,ユーザが確認して必要に応じて修正しなければならない.これら二点への対応が今後の課題である.
なお,本プログラムについてはSwissParamおよびAntechamberから取得したパラメータで動作することを確認済みである.
本プログラムはo2pという名称でhttp://o2p.dip.jpにおいて公開している.
本研究は日本学術振興会 科学研究費 基盤研究(C) 16K07714の支援で実施した.