Abstract
The surface adsorption model calculation database, which we are constructing from systematic quantum chemical calculations, is reported. The database can be utilized to analyze heterogeneous catalytic reactions. As an application, we predicted the experimental catalytic activity for the methane steam reforming reaction with this database by using the sparse modeling techniques.
Recent advances in the data-science methods have drawn attention to high-throughput quantum chemical calculations. In this study, we will report the surface model calculation database, which we are constructing by systematically carrying out the calculations of various chemical species adsorbed on metal surface models and storing their results, for heterogeneous catalytic reaction analyses. As an application example of this database, we attempted to evaluate the catalytic activity (namely, conversion rate) obtained from experiments for the following methane steam reforming (MSR) reaction,
CH4 + H2O → CO + 3H2,
by analyzing the information from the database and to specify its activation factor.
Our script program realized semi-automatic and systematic quantum chemical calculations with SIESTA program [1]. The current database collects the results for the three-layer 3×3 fcc (111), (100), and (211) surface slab models (with 15 Å vacuum layer) and these surfaces with adsorbates at GGA-PBE/DZP level of calculations. Doping effect can be considered by substituting the central atom in the first layer with dopant. For each surface-adsorbate pair, various adsorption structures were considered by randomly generating 100 initial structures except for single-atom adsorption cases. Up to now, 32,454 calculation results are collected in this database.
For the analysis of MSR reaction, we recollected 1,028 calculation results, namely, those for clear surface and adsorption structures of H, C, O, H2, CH, OH, C2, and CO, for each metal surface from the database. As the statistical analysis, we utilized sparse modeling methods, where the following equation (1) for the least square estimation with the regularization term p is minimized to perform the descriptor selection and regression simultaneously:
| l(β)=(y−Xβ)T(y−Xβ)+p(β) | (1) |
. Here,
y represents the objective variable vector (i.e., methane conversion rate),
X is the descriptor matrix, and
β is the regression coefficient vector for descriptors. Namely, the following regularization terms (2) and (3) are used for LASSO [
2] and MC
+ [
3] regressions, respectively:
,
| p(β)=λ∑j∫0|βj|{1−xvλ}+dx | (3) |
where {
x}
+ = max(
x,0). The regularization parameter
λ was determined with the leave-one-out cross validation. These regressions were performed with the ncvreg package [
4] in R language [
5].
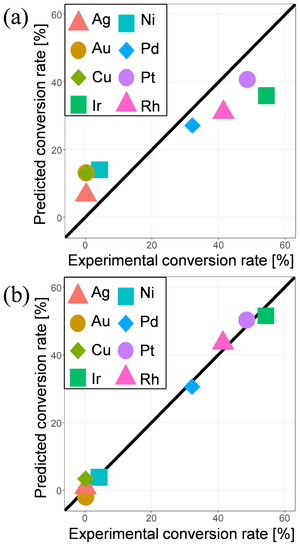
As the objective variable, the experimental conversion rates of methane on clean metal surface at around 1120 K were taken from Ref [6]. Because the rates were obtained from the single article, the experimental conditions other than metal were the same. Table 1 lists the descriptors used in the sparse modeling regression taken from the surface adsorption model database. Since many adsorption structures are collected for adsorption calculations, we took maximum, minimum, average (avg), and standard deviation (sd) of these properties as the descriptors. Figure 1 shows the comparison of the methane conversion rates predicted by LASSO and MC+ methods with the experimental value. Particularly the MC+ prediction shows good agreement with the experimental value. Non-zero regression coefficients by MC+ method are listed in Table 2. It was found that the C2 adsorption energy on (111) surface negatively correlates with the experimental conversion rate, although C2 does not directly appear in the MSR reaction. Note that a negative adsorption energy represents the stabilization by the adsorption and that the C2 adsorption energy on (111) surface was selected also by LASSO regression with the largest negative coefficient. This correlation may reflect the deactivation by carbon deposition.
Table 1. Descriptor candidates taken from the surface adsorption model database to regress the methane conversion rate.
| From metal surface calculation |
| - Relaxation energy for first layer |
| - Average charge of first-layer surface |
| From adsorption calculation for adsorbate X |
| - Adsorption energy (max, min, avg, sd) |
| - Average charge of surface (max, min, avg, sd) |
| - Charge of adsorbate (max, min, avg, sd) |
| - Height of adsorbate (max, min, avg, sd) |
| - Distance b/w adsorbed atoms (max, min, avg, sd) |
Table 2. Descriptors selected by MC
+ method and their regression coefficients (in brackets) for methane conversion rate.
| Max of C2 adsorption energy on (111) surface [-26.54] |
| Min of C2 adsorption height on (111) surface [8.83] |
| SD of C charge for CH adsorption on (111) surface [-1.40] |
The present report shows the usefulness of our surface adsorption model calculation database for the analysis of heterogeneous catalytic reactions. Note that this database can be used not only for the specific MSR reaction but also for wide varieties of heterogeneous catalytic reactions that proceed on metal surfaces. The application of the present method to the experimental results from multiple articles is now under consideration.
Acknowledgment
We are grateful for financial support from the programs of the Ministry of Education, Culture, Sports, Science and Technology (MEXT, Japan) "Elements Strategy Initiative to Form Core Research Center" (Grant Number JPMXP0112101003) and "Priority Issue on Post-K computer" (Development of new fundamental technologies for high-efficiency energy creation, conversion/storage and use). M.K. was supported by JST for PRESTO (Grant Number JPMJPR15N3). Some of the present calculations were performed using the computer facilities at Research Center for Computational Science, Okazaki and at Research Institute for Information Technology, Kyushu University, Japan.
References
- [1]
J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón, D. Sánchez-Portal, J. Phys. Condens. Matter, 14, 2745 (2002). doi:10.1088/0953-8984/14/11/302
- [2]
R. Tibshirani, J. R. Stat. Soc. B, 58, 267 (1996). doi:10.1111/j.2517-6161.1996.tb02080.x
- [3]
C. H. Zhang, Ann. Stat., 38, 894 (2010). doi:10.1214/09-AOS729
- [4]
P. Breheny, J. Huang, Ann. Appl. Stat., 5, 232 (2011). doi:10.1214/10-AOAS388 PMID:22081779
- [5]
R Core Team, "R: A Language and Environment for Statistical Computing," Foundation for Statistical Computing, Vienna, Austria, 2017.
- [6]
D. Mei, V. A. Glezakou, V. Lebarbier, L. Kovarik, H. Wan, K. O. Albrecht, M. Gerber, R. Rousseau, R. A. Dagle, J. Catal., 316, 11 (2014). doi:10.1016/j.jcat.2014.04.021