Introduction
Common buckwheat (Fagopyrum esculentum. Moench) is an ancient pseudo-cereal crop that belongs to the eudicot family Polygonaceae, widely cultivated in temperate regions of Asia, Europe and North America, particularly China, Japan, Korea, Ukraine, Russia, Nepal and Bhutan (Ohnishi 1998a, 1998b). Due to its drought resistance and short growing season, it is widely cultivated in plateau regions including northwest and southwest China (Zhang et al. 2017).
Historically, common buckwheat seed had been used to make noodles, dumpling wrap, pancakes and other products as a healthy diet source, which contains favorable amino acid composition, dietary fibers, trace elements, vitamins (B3, B6 and K), starch and essential minerals (Bonafaccia et al. 2003a, 2003b, Lee et al. 2010). Recently, many researchers reported that common buckwheat seed has considerable economic importance for its pharmaceutical potential to decrease risk of diabetes (Stringer et al. 2013), cancer and cardiovascular disease (Tomotake et al. 2006). Hence, massive studies have mainly focused on biologically active components of common buckwheat grains, such as flavonoids and flavones (Li et al. 2010, Matsui et al. 2018), tannins (Steadman et al. 2001), phytosterols (Krkošková and Mrazova 2005) and fagopyrins (Eguchi et al. 2009, Stojilkovski et al. 2013). Very little research has been done on the yield of common buckwheat despite the high nutritional and nutraceutical value of its seed.
Seed size is one of the main factors affecting seed yield. Transcription factors play important roles in controlling seed size in Arabidopsis thaliana, such as APETALA2 (AP2) (Ohto et al. 2005, 2009) and TRANSPARENT TESTA GLABRA2 (TTG2) encoding a WRKY TF (Johnson et al. 2002). In recent years, several signaling pathways regulating seed size have been identified in Arabidopsis thaliana and Oryza sativa, such as the IKU pathway (Luo et al. 2005, Wang et al. 2010, Zhou et al. 2009b), the ubiquitin–proteasome pathway (Du et al. 2014, Li et al. 2008, Xia et al. 2013), the MAPK signaling pathway (Duan et al. 2014, Liu et al. 2015) and phytohormones (Li et al. 2011, Xu et al. 2015, Zhang et al. 2012a).
An increasing number of studies have confirmed that RNA sequencing (RNA-seq) is a powerful method to investigate the transcriptome profiles and molecular basis of many agronomically important traits (Dussert et al. 2013, Xue et al. 2017, Yin et al. 2013, Zhang et al. 2012b). In common buckwheat, Yokosho et al. (2014) conducted a genome-wide transcriptome analysis of Al-responsive genes in roots and leaves by RNA sequencing (RNA-Seq). Xu et al. (2017) provided insight into Al toxicity and resistance mechanisms in common buckwheat root apices through transcriptome analysis of Al-reduced genes. Shi et al. (2017) firstly analyzed the immature seed transcriptome of common buckwheat and developed genic-SSR markers.
Nevertheless, the mechanism of seed development of common buckwheat remains unclear at the molecular level and no genes related to seed size have been identified. In this study, RNA-Seq was used to analyze the transcriptomes of seed at two development stages in two cultivars of common buckwheat (large-seeded and small-seeded). This transcriptome dataset could provide useful information to understand the molecular mechanism of seed development, and reveal potential genes related to seed size in common buckwheat.
Materials and Methods
Plant materials
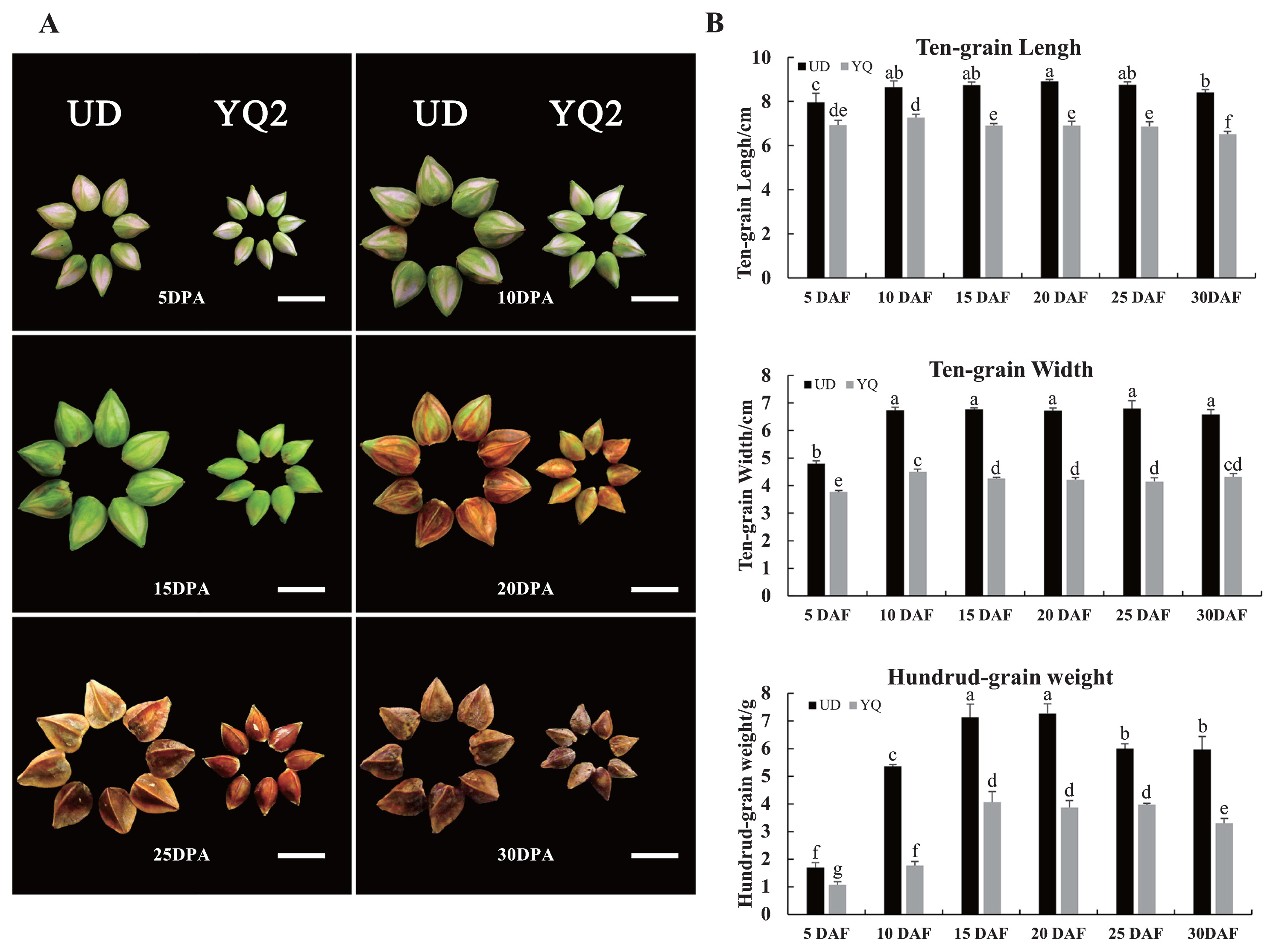
Two typical cultivars of common buckwheat, Youqiao2 (YQ) (small-seeded) and Ukraine daliqiao (UD) (large-seeded), were planted in the test field at Southwest University, Chongqing, China in spring, 2017, with normal field management during the growth period. Seeds of 5, 10, 15, 20, 25 and 30 day s post anthesis (DPA) were collected from YQ and UD for morphological observation of seed growth. Based on the morphology of the seeds, samples for RNA-Seq were collected at 5 and 10 DPA. For RNA extraction, the materials were frozen in liquid nitrogen immediately and stored at −80°C.
RNA extraction, library construction and sequencing
Total RNA was extracted using TRNzol Reagent (TIANGEN, Beijing, China), according to the manufacturer’s instructions. The purity and contamination of the isolated RNA was monitored on 1% agarose gels and Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, USA). We used the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA) to assess RNA integrity. Subsequently, these RNA samples were used for cDNA library construction, respectively.
For library construction, equal quantities (1.5 μg) of total RNA per sample were used as input for RNA sample preparations. Sequencing libraries were generated using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer’s instructions. In brief, poly-T oligo-attached magnetic beads were used to purify mRNA from total RNA. We used random hexamer primers and M-MuLV Reverse Transcriptase (RNase H) to synthesize first strand cDNA. Subsequently, second strand cDNA synthesis was performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. In order to select cDNA fragments of 150~200 bp in length, the library fragments were purified, size-selected, and adaptor-ligated. Then PCR was performed and PCR products were purified to create the final cDNA libraries. Library quality was assessed on the Agilent Bioanalyzer 2100 system.
The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina) according to the manufacturer’s instructions. After cluster generation, the libraries were sequenced on an Illumina Hiseq platform and paired-end reads were generated by Novogene Bioinformatics Technology Co. Ltd., Beijing, China (www.novogene.cn).
Sequence assembly and annotation
Raw data (raw reads) in FASTQ format were first processed through in-house perl scripts. Clean data (clean reads) were obtained by removing reads containing adapter, reads containing poly-N and low quality reads from raw data. Transcriptome assembly was accomplished using Trinity software (Grabherr et al. 2011) with min_kmer_cov set to 2 by default and all other parameters set to default. Unigenes of the transcriptome were annotated based on data from the Nr (NCBI non-redundant protein sequences), Nt (NCBI non-redundant nucleotide sequences), Pfam (Protein family), KOG/COG (Clusters of Orthologous Groups of proteins), Swiss-Prot (manually annotated and reviewed protein sequence), KO (KEGG Ortholog), and GO (Gene Ontology) databases. The COGs protein database phylogenetically classifies the complete complement of proteins encoded in a genome. Each COG is a group of three or more proteins that are inferred to be orthologs or direct evolutionary counterparts. To further analyze the transcriptome of F. esculentum, all unigenes were submitted to the KEGG pathway database. All BLAST searches were performed with an e-value of 1E−5.
Analysis of differentially expressed genes
Gene expression levels were estimated by RSEM (Li and Dewey 2011) for each sample. Clean data were mapped back onto the assembled transcriptome. Read count for each gene was obtained from the mapping results. Prior to differential gene expression analysis, for each sequenced library, read counts were adjusted by edgeR program package through one scaling normalized factor. Differential expression analysis of two samples was performed using the DEGseq (2010) R package. P-value was adjusted using q value (Storey and Tibshirani 2003). Q value < 0.005 & |log2(foldchange)| > 1 was set as the threshold for significant differential expression.
Validation of differentially expressed genes by qRT-PCR analysis
After RNA-seq, qRT-PCR was performed by SYBR Premix Ex Taq II (Tli RNaseH Plus) in Bio-Rad CFX manager 2.0. Details of the primers used for the qRT-qPCR assay were listed in Supplemental Table 1. H3 was used as an internal control to verify real-time quantitative PCR. The values from three independent biological replicates and three technical replicates were averaged. The Ct values were determined and compared with the 2−ΔΔCt method (Livak and Schmittgen 2001).
Results
Morphological observation of seed growth on common buckwheat
The growth status and appearance of seeds of UD and YQ were shown in Fig. 1. Before 10 DPA, the seed coat had rapid growth, almost to the maximum at 10 DPA. From 10 to 20 DPA, no obvious change in seed coat size was found. Seeds plumped up little by little and seed contents increased as well. Most nutrients accumulated at these stages. After 20 DPA, the seed coat turned brown and seeds become slightly lighter after dehydrating, indicating that seeds were fully mature (Fig. 1A). During the whole growing period, the grains of UD were significantly larger than those of YQ and the key period determining seed size was before 10 DPA in common buckwheat (Fig. 1B).
RNA sequencing and gene annotation
To investigate the underlying mechanisms that control seed size of common buckwheat, high-throughput RNA-Seq was performed using seeds at 5 DPA and 10 DPA from UD (large-seed) and YQ (small-seed), respectively. Four libraries were constructed and sequenced using an Illumina HiSeq™ 2000 platform. Totals of 54,002,588, 51,853,908, 48,487,090 and 47,547,216 raw reads were generated. The raw reads are available in the NCBI Sequence Read Archive (SRA accession: PRJNA523295). After stringent quality assessment and data filtering, totals of 52,697,868, 50,423,336, 47,398,614 and 45,832,030 clean reads were generated for UD_5 (5 DPA seed), UD_10 (10 DPA seed), YQ_5 (5 DPA seed), YQ_10 (10 DPA seed), respectively. A total of 259,895 transcripts were assembled (Table 1). Transcript lengths ranged from 201 to 14,557 bp. The de novo assembled transcriptomes were clustered and the longest clusters were defined as unigenes. A total of 187,034 unigenes were obtained from transcripts with average length of 1097 bp and N50 of 1538 bp (Table 1).
Table 1
Characteristics of assembled common buckwheat transcripts and unigenes
| Nucleotide length (bp) |
Transcripts |
Unigenes |
| <301 |
61787 |
15768 |
| 301–500 |
58341 |
36590 |
| 501–1000 |
63855 |
59293 |
| 1001–2000 |
50534 |
50028 |
| >2000 |
25378 |
25355 |
| Total |
259895 |
187034 |
| Min length (bp) |
201 |
201 |
| Mean length (bp) |
878 |
1097 |
| Max length (bp) |
14557 |
14557 |
| N50 (bp) |
1381 |
1538 |
| N90 (bp) |
363 |
523 |
| Total nucleotides (bp) |
228,079,666 |
205,214,595 |
To identify the putative functions of the assembled unigenes, all were compared to sequences in Nr, Nt, Ko, Swiss-Prot, Pfam, GO, KOG and KEGG public databases, (Table 2). Among 187,034 unigenes, 20,349 (10.88%) unigenes were annotated in all databases, and 138,101 (73.84%) were annotated in at least one of the above public databases. A total of 128,444 (68.67%) unigenes matched sequences in the Nr databases.
Table 2
Functional annotation of the grain transcriptome in
F. esculentum
|
Number of unigenes |
Percentage (%) |
| Annotated in NR |
128444 |
68.67 |
| Annotated in NT |
66181 |
35.38 |
| Annotated in KO |
53770 |
28.74 |
| Annotated in Swiss-Prot |
99269 |
53.07 |
| Annotated in PFAM |
89160 |
47.67 |
| Annotated in GO |
90622 |
48.45 |
| Annotated in KOG |
34060 |
18.21 |
| Annotated in all databases |
16366 |
8.75 |
| Annotated in at least one database |
138101 |
73.83 |
| Total unigenes |
187034 |
100 |
In the GO functional annotation classification system, 187,034 unigenes were classified into three main categories: biological processes (47.59%), cell composition (29.04%) and molecular function (23.39%). There were 25, 21 and 10 functional categories within the three main categories, respectively (Supplemental Fig. 1A). For the biological process category, many high abundance unigenes were classified into “cellular process”, “metabolic process” and “single-organism process”. In the cellular component category, many unigenes were categorized as “cell” and “cell part”. Under the molecular function category, “binding” and “catalytic activity” represented major categories.
In the KOG functional classification system, 34,060 unigenes obtained in our study were classified into 25 categories (Supplemental Fig. 1B). The largest group was “posttranslational modification, protein turnover, chaperones” (4,628; 13.59%), followed by “general function prediction only” (4,358; 12.8%); “translation, ribosomal structure and biogenesis” (3,301; 9.7%); “Intracellular trafficking, secretion, and vesicular transport” (2,500; 7.34); “Signal transduction mechanisms” (2,491; 7.31%); “RNA processing and modification” (2,418; 7.1%).
To further identify the biological pathways of F. esculentum seed unigenes, a total of 42,142 unigenes were classified into 5 main categories including 130 KEGG pathways (Supplemental Fig. 1C). “Metabolism” accounted for 57.48% (24,224), was the biggest category, followed by “Genetic Information Processing” (11,249; 26.69%), “Cellular Processes” (2,728; 6.47%), “Environmental Information Processing” (2,118; 5.02%), “Organismal Systems” (1,823; 4.32%). The F. esculentum seeds had high carbon metabolism (Ko01200, 1966), biosynthesis of amino acids (Ko01230, 1700), protein processing in endoplasmic reticulum (Ko04141, 1428), starch and sucrose metabolism (Ko00500, 1397) and plant hormone signal transduction (Ko04075, 1317). In addition, a few unigenes were distributed to indole alkaloid biosynthesis (Ko00901, 1) and isoflavonoid biosynthesis (Ko00943, 9).
Differentially expressed gene (DEG) identification and functional annotation
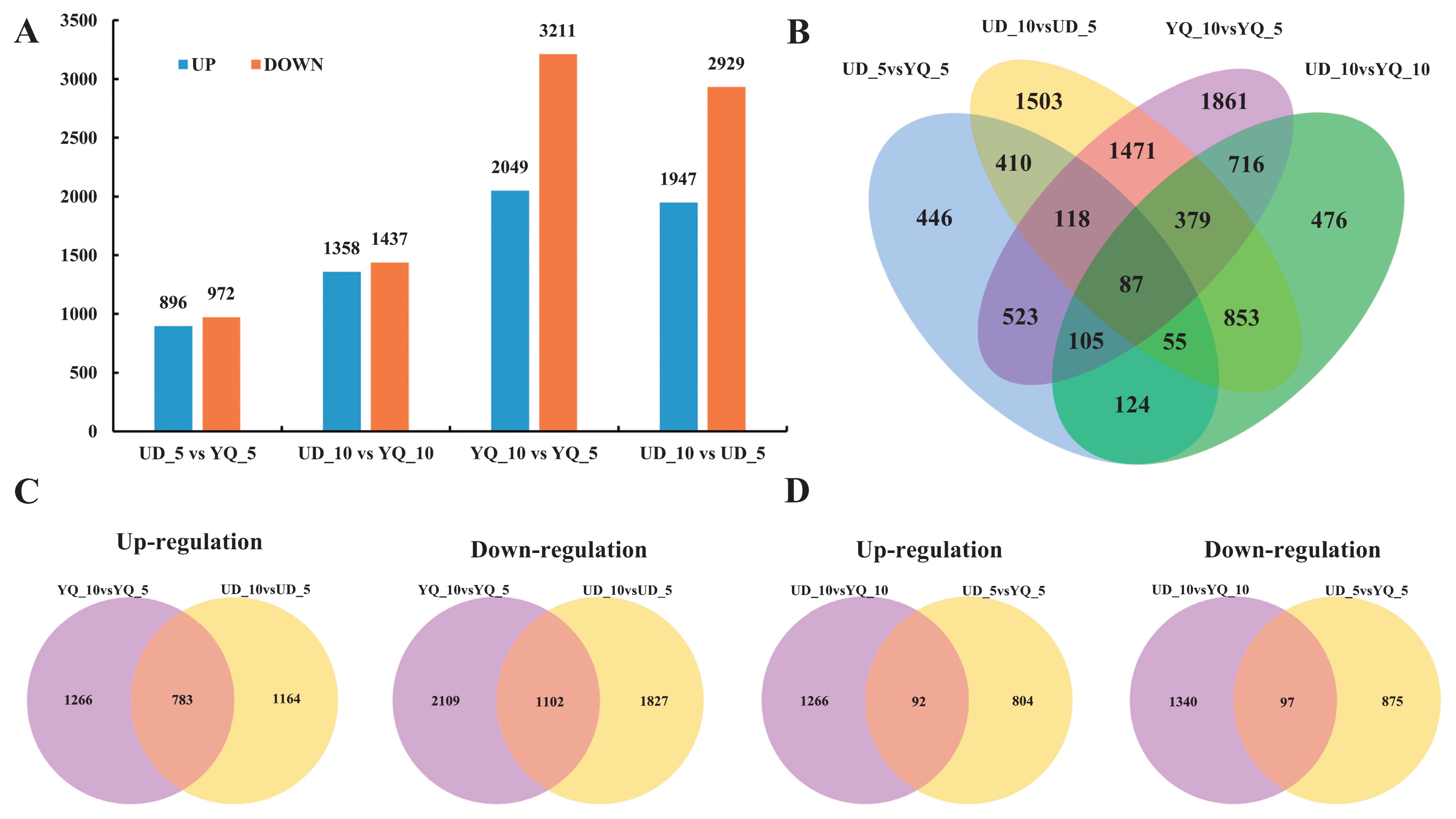
To explore potential differential expression of genes between four samples, clean reads were mapped to the unigene database. The expression levels of unigenes were measured by FPKM (expected number of Fragments Per Kilobase of transcript sequence per Million base pairs sequenced) values. In this study, a total of 9127 DEGs were detected at two stages of seed development in the two cultivars (UD_5, UD_10, YQ_5 and YQ_10). In the UD_5 vs. YQ_5, UD_10 vs. YQ_10, YQ_10 vs. YQ_5 and UD_10 vs. UD_5, we identified 1868, 2795, 5260 and 4876 DEGs, respectively (Fig. 2A). Among these DEGs, 87 were differentially expressed in all comparisons (Fig. 2B), with 783 up-regulated and 1102 down-regulated genes detected simultaneously in the UD_10 vs. UD_5 and YQ_10 vs. YQ_5 comparisons. Totals of 92 up-regulated and 97 down-regulated genes were detected simultaneously in the UD_5 vs. YQ_5 and UD_10 vs. YQ_10 (Fig. 2C, 2D).
The GO enrichment analysis of DEGs was performed using four pairwise comparisons (Supplemental Table 2). “Metabolic process” was enriched with the most DEGs, except UD_10 vs. UD_5, in which most DEGs were assigned to “single-organism process”. Nevertheless, “ribosome”, “ribosome biogenesis”, “structural constituent of ribosome” and “ribonucleoprotein complex biogenesis” were the most significantly enriched GO terms in UD_5 vs. YQ_5 and UD_10 vs. YQ_10. “Nutrient reservoir activity”, “single-organism metabolic process”, “oxidation-reduction process”, “oxidoreductase activity” and “carbohydrate metabolic process” were significantly enriched in UD_10 vs. UD_5 and YQ_10 vs. YQ_5.
To further understand the metabolic pathways in which DEGs were involved, KEGG analyses were performed (Supplemental Table 3). In comparing UD_5 vs. YQ_5, DEGs were significantly enriched in seven KEGG pathways, including “Ribosome”, “Flavonoid biosynthesis”, “Phenylpropanoid biosynthesis”, “Phenylalanine metabolism”, “Carbon fixation in photosynthetic organisms”, “Circadian rhythm - plant” (corrected P-value < 0.05). In UD_10 vs. YQ_10, only “Ribosome” and “Protein processing in endoplasmic reticulum” were significantly enriched in DEGs. Five and eight KEGG pathways were significantly enriched in DEGs of UD_10 vs. UD_5 and YQ_10 vs. YQ_5, respectively. Interestingly, “Pentose and glucuronate interconversions” and “phenylpropanoid biosynthesis” were significantly enriched consistently in UD_10 vs. UD_5 and YQ_10 vs. YQ_5.
Identification of differentially expressed transcription factors
Transcription factors (TFs) play an important role in the growth of different seed tissues by either enhancing or repressing cell division and expansion. In this study, a total of 6856 genes belonging to 80 TF families, were identified. Among these, 545 TFs from 62 families showed significant differential expression in one or more comparisons (Fig. 3A). Among the 62 TF families, AP2-EREBP, ARF, MYB, bZIP and WRKY TF families had a relatively high proportion of differentially expressed genes, which had been previously shown to have important roles in regulating seed size in Arabidopsis (Fig. 3A).
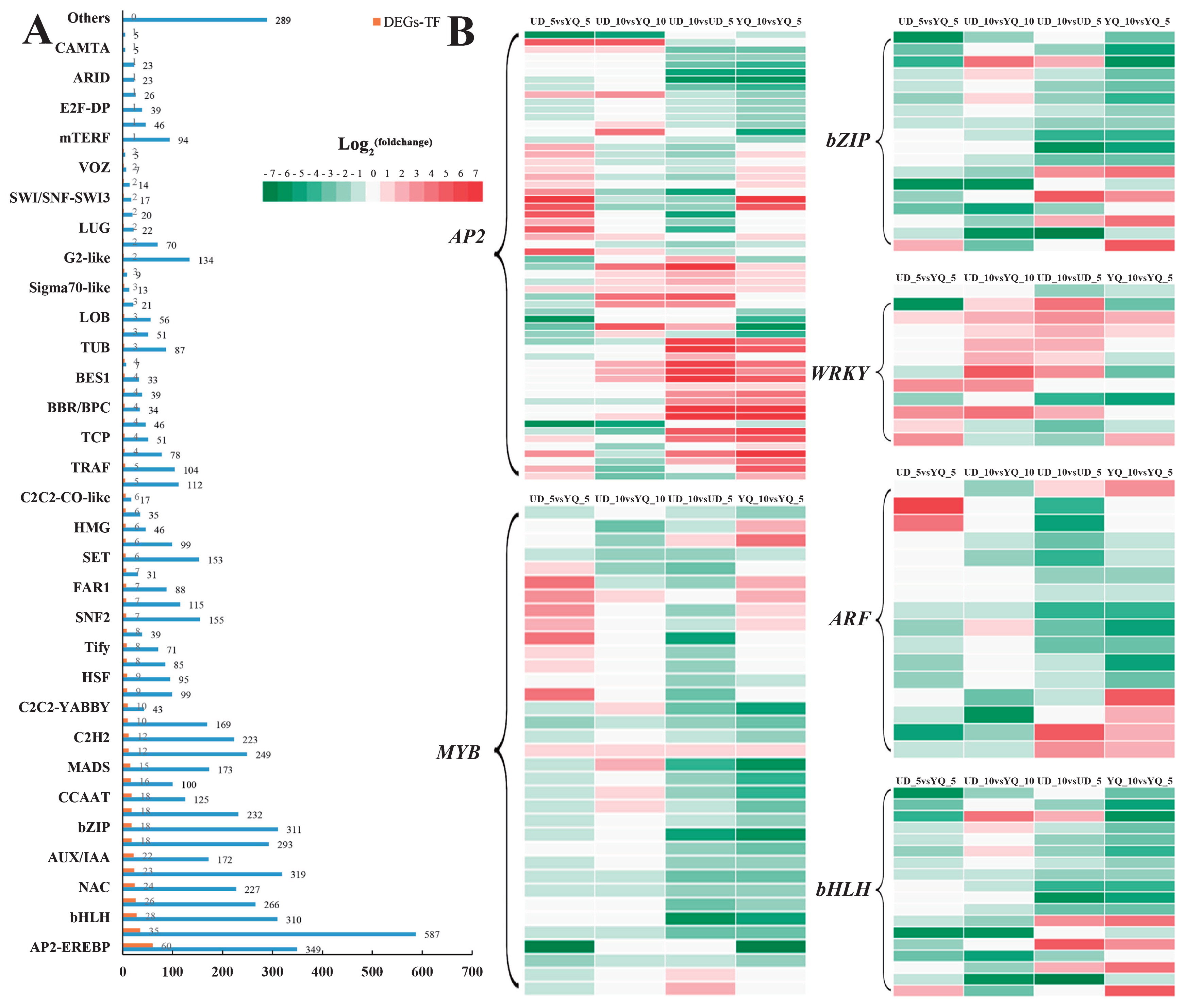
In this study, we identified 349 DEGs from six TF families, including 60 AP2, 16 ARF, 18 bZIP, 28 bHLH, 35 MYB and 12 WRKY (Fig. 3B, Supplemental Table 4). For the UD_10 vs. UD_5 and YQ_10 vs. YQ_5 comparisons, most DEGs in ARF, MYB, bHLH and bZIP TF families were down-regulated with seed development. For the UD vs. YQ comparison, most DEGs in AP2, MYB, bHLH and WRKY TF families were up-regulated in UD. All DEGs in bZIP were down-regulated in UD. Among them, five DEGs were detected simultaneously in UD_5 vs. YQ_5 and UD_10 vs. YQ_10 comparisons. Two AP2 (Cluster-3342.81142, Cluster-3342.101865) and one WRKY (Cluster-3342.67464) were significantly up-regulated, whereas two bZIP (Cluster-3342. 116302, Cluster-3342.84135) were significantly down-regulated in UD vs. YQ.
Identification of differentially expressed genes involved in the ubiquitin–proteasome pathway
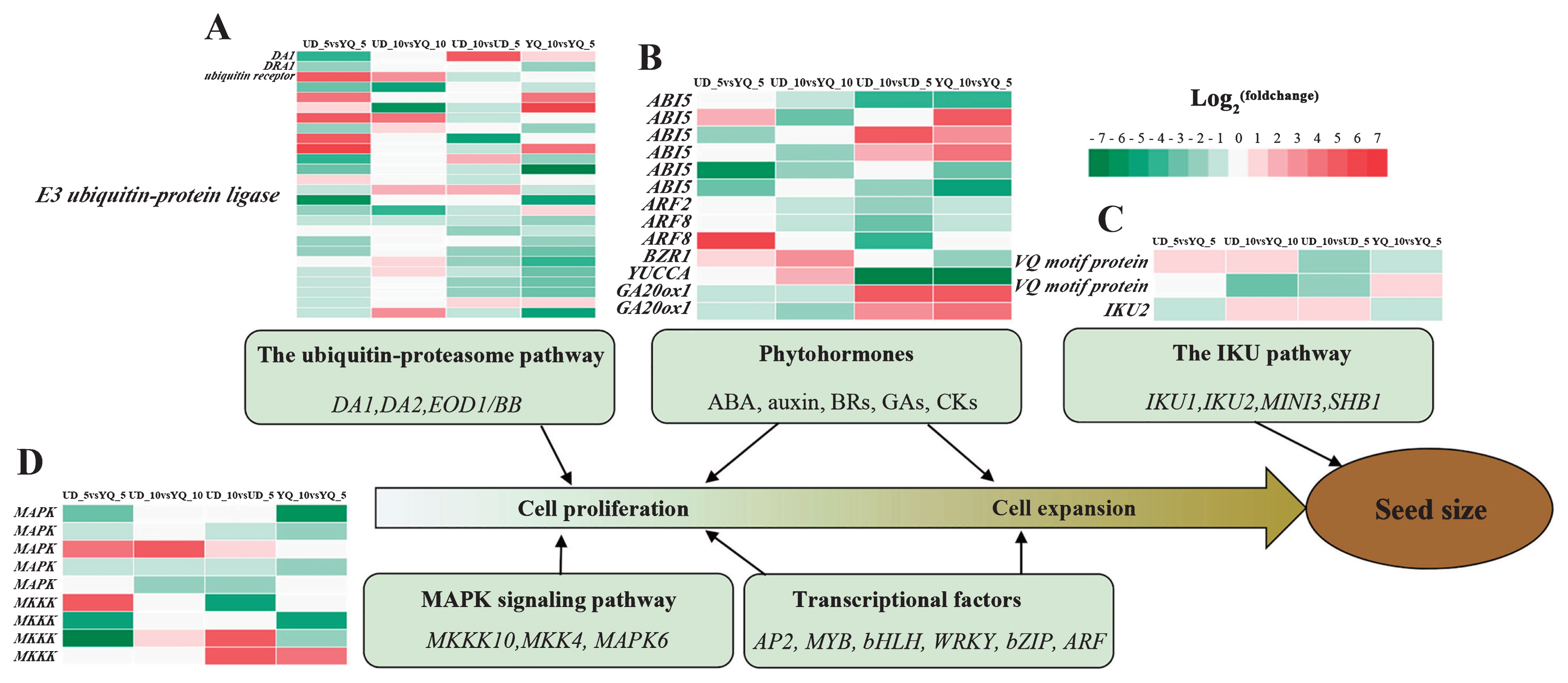
In the ubiquitin–proteasome pathway, we identified 25 DA1, 23 DAR1 (DA1-related 1), 22 DAR2 (DA1-related 2) and two BB (BIG BROTHER), but no gene showed significant differential expression in four comparisons (Supplemental Table 5). In addition, we identified 13 genes annotated as ubiquitin receptor and 684 annotated as E3 ubiquitin-protein ligase in this study, including one and 25 DEGs, respectively (Supplemental Table 5, Fig. 4A). A total of 19 DEGs, annotated as E3 ubiquitin-protein ligase, were identified in the UD_10 vs. UD_5 or YQ_10 vs. YQ_5 comparisons and most were down-regulated in seed development, which implied their vital regulatory mechanisms in the seed development of common buckwheat.
Identification of differentially expressed genes involved in phytohormones
Plant hormones regulate diverse processes in growth and development. In this study, a total of 151 unigenes were identified among six types of phytohormones known to function in regulating seed size in Arabidopsis thaliana or rice, including ABSCISIC ACID-INSENSITIVE5 (ABI5, 44), Auxin response factor 2 (ARF2, 19), Auxin response factor 8 (ARF8, 19), Auxin response factor 19 (ARF19, 7), BRASSINAZOLE-RESISTANT 1 (BZR1, 5), BRASSINOSTEROID INSENSITIVE 1 (BRI1, 35), YUCCA (YUC, 14), GA20ox1 (4) and cytokinin dehydrogenase (CKX, 4) (Supplemental Table 5). Among these, 13 DEGs were identified, five in UD vs. YQ comparisons and 11 in 10 DPA vs. 5 DPA comparisons (Fig. 4B). Three ABI 5 and two GA20ox1 were significantly up-regulated but others DEGs were significantly down-regulated with seed development. This result implies critical functions of these genes in controlling seed development of common buckwheat.
Identification of differentially expressed genes involved in IKU pathway
In the IKU pathway, we identified 26 unigenes, including four encoding VQ motif-containing proteins, 17 HAIKU2 genes, four encoding EXS family proteins and one encoding SPX domain-containing protein (Supplemental Table 5). Among these, three DEGs were identified (Fig. 4C). Two genes encoding VQ motif-containing protein exhibited different expression patterns: cluster-3342.64210 was up-regulated at the 5 and 10 DPA stages in UD compared with YQ, especially in 5 DPA, whereas cluster-3342.71829 was up-regulated at 5 DPA and followed by significant down-regulation at 10 DPA in UD compared with YQ. One HAIKU2, cluster-3342.65038, was identified, whose expression level was down-regulated at 5 DPA but later down-regulated at 10 DPA in UD compared with YQ.
Identification of differentially expressed genes involved in MAPK signaling pathway
In the MAPK signaling pathway, 97 MAPK, 21 MAPKK and 123 MKKK unigenes were identified, among which only nine were DEGs, five MAPK and four MKKK (Supplemental Table 5, Fig. 4D). One MAPK (cluster-3342.67812) showed up-regulation in the four comparisons, whereas another four MAPK were down-regulated in seed development. Four MKKK showed opposite expression patterns: parts were up-expressed and the rest were down-expressed, suggesting that they may have different functions in regulating seed development of common buckwheat.
qRT-PCR validation of DEGs from the RNA-Seq analysis
To validate the expression patterns of DEGs obtained from RNA-Seq analysis, qRT-PCR was conducted to examine the expression levels of 16 DEGs in the two development stages on two cultivars (Fig. 5A). The expression levels of these selected genes from qRT-PCR analyses were generally consistent with FPKM values deduced from RNA-Seq (Fig. 5A). The correlation between qRT-PCR and RNA-Seq measurements showed a coefficient of determination (R-squared) of 0.843 (Fig. 5B). These results confirm the reliability of the transcriptomic profiling data estimated from RNA-Seq data.
Discussion
Seed development starts with double fertilization, in which one male gamete fertilizes a haploid egg cell to form an embryo and the second male gamete fertilizes a diploid central cell to form the triploid endosperm. (Li and Li 2014, Savadi 2018). Therefore, a seed mainly consisted of three parts, the embryo, endosperm and seed coat (integuments), biologically (Savadi 2018). In this study, we found that the seed coat had rapid growth before 10 DPA, almost reaching maximum size at 10 DPA. There was no obvious change of grain width and length after 10 DPA (Fig. 1B), which suggested that the seed coat sets an upper limit to final seed size. A similar pattern had been observed in rice, where the spikelet hull may set the final size of a grain (Song et al. 2007, Zhou et al. 2009a). Therefore, we speculated that the seed coat limits the seed size of YQ (small-seeds) and could be responsible for the difference of seed size between UD and YQ.
To investigate differential expression of genes related to seed size in common buckwheat, we performed genomewide transcriptome sequencing using 5 DPA and 10 DPA seed from two cultivars and identified 187,034 unigenes. Among these, 6856 TF genes were identified, including 545 DEGs in one or more than one comparison(s). Most of these DEG-TFs were members of several TF families known to function in regulating seed size in Arabidopsis thaliana, such as AP2-EREBP (Jofuku et al. 2005, Ohto et al. 2005, 2009), ARF (Goetz et al. 2006, Hughes et al. 2008, Schruff et al. 2006), MYB (Gonzalez et al. 2009, Ishida et al. 2007, Li et al. 2009), bHLH (Ishida et al. 2007), bZIP (Cheng et al. 2014) and WRKY (Luo et al. 2005). The largest was the AP2-EREBP TF family, which had been demonstrated genetically to determine seed size and seed weight by coordinating the growth of maternal integuments, endosperm and embryo (Jofuku et al. 2005, Ohto et al. 2005, 2009). In this study, we identified 381 AP2, among which 22 and 16 AP2 showed differential expression in UD_5 vs. YQ_5 and UD_10 vs. YQ_10, respectively (Supplemental Table 5). Interestingly, two AP2 (cluster-3342.81142, cluster-3342.101865) had significantly up-regulated expression in UD_5 vs. YQ_5 and UD_10 vs. YQ_10, simultaneously, demonstrating that AP2 could play an important role in controlling seed size of common buckwheat.
In previous studies, the expression level of AP2 decreased by BR treatment and increased by BRZ treatment (Jiang et al. 2013). Overexpression of OsBZR1, a BR-signaling transcription factor, resulted in higher grain yield in rice (Zhu et al. 2015). In this study, we identified five BZR1 homologs. One homolog of BZR1 (cluster-3342.73464) was simultaneously up-regulated in UD_5 vs. YQ_5 and UD_10 vs. YQ_10 comparisons. We speculated that BR-signal transduction was involved in controlling seed size by regulating the expression level of AP2 in common buckwheat.
Except for brassinosteroids, ABA had been reported to regulate early seed development in Arabidopsis (Cheng et al. 2014). The bZIP transcription factor ABI5, involved in ABA signaling, negatively mediates seed size (Cheng et al. 2014), which was concordant with one of the interesting findings in this study, that two ABI5 genes had significantly down-regulated expression in UD (large-seeded), compared with YQ (small-seeded). This result possibly indicated that bZIP transcription factor ABI5 could be another key gene controlling seed size of common buckwheat.
The ubiquitin–proteasome pathway has recently been shown to regulate seed size. The ubiquitin receptor DA1 and DA1-related protein (DAR1) sets final seed and organ size by restricting cell proliferation in maternal integuments (Li et al. 2008). In this study, we identified 25 DA1, 23 DAR1, 22 DAR2, but there were no genes showing significant differential expression between UD and YQ. This finding implied that the seed size difference between UD and YQ was likely attributable to cell expansion, not cell proliferation of maternal integuments.
In the present study, putative orthologs involved in the IKU pathway showed no significant differences in expression between UD and YQ. This result was likely because these genes in the IKU pathway governed seed size by influencing endosperm growth rather than the maternal integuments (Luo et al. 2005, Wang et al. 2010).
The MAPK signaling pathway has been characterized to play important roles in controlling grain size in rice (Guo et al. 2018, Xu et al. 2018). In this study, we identified 97 MAPK, 21 MAPKK and 123 MAPKKK genes but the lack of MAPK signaling pathway unigenes showing significant differences impels further study to investigate their exact functions in seed development of common buckwheat.
qRT-PCR has been frequently applied to validate expression levels of specific genes obtained by RNA-Seq, such as expression levels detected at time-points that were generally consistent with mRNA-Seq findings (Hacquard et al. 2010, Huang et al. 2017, Rincão et al. 2018, Tremblay et al. 2008). In the present study, the observed expression patterns of the 16 genes selected from RNA-Seq data were very similar to those obtained for qRT-PCR analyses. Although there is no biological duplication in transcriptome data, three biological replications were performed by qRT-PCR, and the expression levels of these selected genes from qRT-PCR analyses were generally consistent with those deduced from RNA-Seq, indicating that transcriptome data obtained in this study can be used in future studies.
The study provides a global transcriptome survey from common buckwheat seeds at two development stages from two cultivars (large-seeded and small-seeded). Focusing on TFs, phytohormones, the IKU pathway, the ubiquitin–proteasome pathway and the MAPK signaling pathway, differentially expressed genes between UD (large-seeds) and YQ (small-seeds) were analyzed and discussed. We proposed that AP2 and bZIP transcription factors, BR-signal and ABA were regulators of seed size of common buckwheat. This transcriptome data of common buckwheat seeds will facilitate further research on seed size and provide a theoretical basis for improving yield of common buckwheat.