SAMPLE PREPARATION
Throughout the sample preparation procedure, special care should be taken not to degrade or de-localize the molecule(s) of interest. For the earliest IMS applications, proteins and peptides were in focus, and hence many sample preparation strategies are formulated with these molecules in mind. Beginners in IMS should take extra care to find or develop sample preparation strategies that suit their tissues or analytes of interest. General recommendations for successful imaging experiments are given below.
Tissue stabilizationThere are useful strategies for hampering the post mortal changes of biological samples. Studies have shown rapidly occurring increases or decreases in abundance for a number of molecules when the samples are kept at room temperature and at normal room humidity.8–10) Stabilization is thus highly recommended and can be performed through heat stabilization, microwave irradiation, formalin fixation or simply through flash freezing. For freezing, the use of powdered dry ice keeps the samples from cracking. Powdered dry ice is easily manufactured by breaking up dry ice with a hammer and separating the resulting powder from the larger pieces of ice through a common sieve.3) For the analysis of rapidly degrading neurotransmitters such as acetylcholine, in situ freezing (ISF) has been shown to ameliorate detectability. For ISF, the model animal is deeply anesthetized and sacrificed by carefully dipping the tip of the head into liquid nitrogen.10) Heat stabilization using the Denator system, where samples are rapidly heated to 95°C, has been proven effective for a number of proteins and peptides.8,11) Preliminary data suggests that heat stabilization is useful also for the analysis of lipids (data not shown). The heat-stabilized tissue may become somewhat fragile when frozen. Resulting difficulties in cryosectioning may be circumvented by utilizing double-adhesive carbon tape, as recently described in a paper by Goodwin et al.9)
Formalin fixation paraffin embedding (FFPE) is a preservation method with a long history in pathology, with millions of clinical samples available in biobanks around the world. Fortunately, sample preparation protocols have been developed that enable the analysis of proteins and peptides in FFPE tissue.12,13) For the analysis of lipids, some species may be analyzed with no special pre-treatment of the samples.14) One should remember that heat stabilization, formalin fixation and microwave irradiation permanently protects samples from enzymatic changes, whereas freezing only preserves the sample state until the temperature is risen and enzymatic processes re-occur.
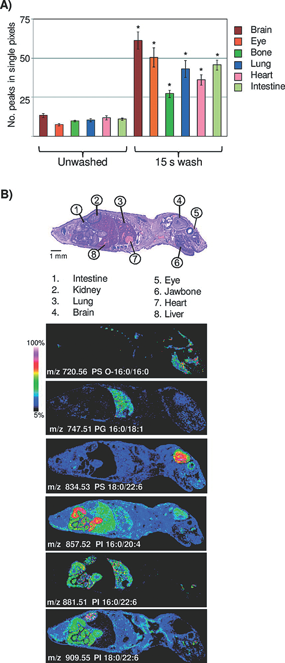
Fig. 1. Aqueous washing illustrated on a whole body mouse pup by MALDI TOF. (A) Comparison of peak number between unwashed and washed tissue. ∗=Student’s t test p-value <2.0×10−4. (B) Examples of images obtained across a whole body mouse pup, illustrating organ specific lipid expression in the negative ion mode. Figure reprint by permission, Angel et al. Anal. Chem. 7(84): 1557–1564, 2012.
Preparation of tissue samples starts with tissue extraction and is followed by tissue embedding, sectioning, and mounting. Post mounting, the samples can either be directly prepared for analysis, or stored at −80°C. Analysis preparation steps for the tissue sections include washing, drying and coating with a carefully selected MALDI matrix. Our most important tips for the cryosectioning are the following; i) Due to the rapid metabolic turnover, work rapidly and in a reproducible manner; ii) Extracted tissue should be frozen or stabilized immediately; iii) Embedding should be performed using a material that does not interfere with mass spectrometry (MS) analysis. We recommend a semi-liquid gel of 2% sodium carboxymethylcellulose (CMC)15) or even a small pool of water. Optimal cutting temperature (OCT) compound or other polymeric materials should be avoided; iv) A tissue thickness of <20 μm improves spectrum quality.3)
WashingWashing procedures should be chosen with the analyte(s) of interest. For peptide or protein analysis, salts and lipids should be removed in order to avoid analytically disturbing ion suppression and hence, the washing solutions will in general contain organic solvents.16,17) These washing solutions are evidently unsuitable for lipid analysis. In addition, organic solvents for tissue washing are often not suitable for the analysis of drug compounds, since drug solubility leads to displacement or even total loss from the tissue. Shariatgorji et al. developed a protocol where the pHs of the washing solutions were adjusted to levels where the drug would have low solubility. Mouse brain tissue treated with one of three different drug compounds (cimetidine, imipramine or “compound c”) had their sections washed with ammonium acetate buffer at pH 10, significantly increasing signal intensities as compared to the results from using acidic or neutral buffers.18) For the analysis of lipids, washing steps are usually avoided. However, Angel et al. developed a washing protocol for the analysis of lipids in negative mode utilizing ammonium formate at pH 6.4 or ammonium acetate at pH 6.7, which significantly increased the number of detectable analytes along with their signal intensities.19)
A third example where washing procedures were specifically developed to fit the analysis is a washing protocol for fragile tissue sections; van Hove et al. developed a method where, instead of immersing the tissue in washing solution, a fiber-free paper tissue was pre-wetted in washing solution and placed on top of the tissue section for 30 to 60 s. If desirable, smaller sections of the tissue can be specifically targeted for this washing procedure.20) Before moving to the matrix application step, the tissue sections should be somewhat dried, either under a swift air of nitrogen gas or in a desiccator for 10–30 min. This process enhances the stability of tissue adhesiveness to the slides in the mass spectrometer.
Matrices and matrix application strategiesAll sample preparation steps are important for high quality imaging results. However, it could be argued that the single most crucial step for successful IMS is the matrix application—both in regards to the choice of matrix, and the tissue coating procedure. There is a plethora of matrices available, all with their individual compound compatibilities, which means that the matrix should be chosen with each analyte at hand.3) New matrices and novel methods for their coating are continuously tested.21–24) There are some practical parameters to keep in mind when choosing matrices. For example, the sublimation rate of your matrix in the vacuum environment of the MS instrument should be investigated; the thickness of the matrix layer should be consistent throughout the analysis, or else the signal intensity is at risk of decreasing over time. In some cases, additives can be used to prolong matrix lifetime or to reduce the effect of alkali adducts.21) In addition, if your matrix peaks overlap with the m/z of your analyte(s), it might be possible to induce a matrix peak mass shift by deuterating the matrix. Thereby, the masking problem is avoided.25) In cases where a non-biased omics approach is employed, a matrix with a low number of matrix peaks is beneficial, to minimize the risk of masking potential biomarkers. Regarding application strategies; there are a number of choices, ranging from manual sprayers to spotting instruments. Any method is satisfactory, as long as it results in homogenous coating and small crystal sizes. Wetting of the tissue should be avoided, since the matrix solvent might de-localize the analytes of interest. When applying matrix through TLC sprayers or air-brushes, the first rounds of spraying should be short in order to create a first layer of seeding crystals. Make sure to wait long enough between the spraying rounds for the tissue to fully dry. Dry matrix can be applied for the analysis of lipids or drugs,26–28) however the results for peptides and proteins have not yet been satisfactory. A large number of publications focus on the different matrices and matrix application strategies,3) and we strongly recommend the beginner in IMS to read in to this particular part of the experiments.
Compound identificationA challenge for the IMS technology compared to other MS technologies is the lack of upstream separation technologies. Due to the extraordinarily large number of molecules present, in combination with the matrix-inherent ion signals, there is an obvious risk of detecting molecules with overlapping m/z values. This in turn leads to a difficulty in ensuring non-ambiguous molecular identification. Fortunately, there are several possible solutions to this problem. For example, the combination of the imaging platform with high-resolution mass analyzers such as orbitrap is one route that has been successfully tested.29) Also, ion mobility is a strategy that offers a post-ionization separation step where isobaric molecules may be distinguished based on their unique molecular cross section area. This approach was successfully used to separate lipid molecules of m/z 746.5 and 746.6, respectively, and illustrate their differing molecular distribution in xenograft breast cancer tissue30) (Fig. 2). A third approach is the use of imaging MS/MS where the distribution images for daughter ions are matched to those of the precursor ion, thereby adding an extra dimension to the confidence in the molecular identification process.10)

Fig. 2. Ion mobility separation of biomolecular ions detected from thin tumor tissue sections. A) Representative spectrum of peak-picked data acquired during an IMS imaging experiment. The lipid peaks are highlighted in red, while matrix-related ions are shown in blue. B) Drift plot of the separated ions. Ion image 1 shows the distribution of a background matrix ion. Ion images 2 (m/z 746.5) and 3 (m/z 746.6) show the distribution of different lipids in the tumor tissue. Lipid-related ions (highlighted in red) were separated from background ions (blue). Figure reprint by permission, Chughtai et al. J. Lip. Res. 54(2): 333–344, 2013.
All IMS data are comprised of x- and y-coordinates, m/z values and ion signal intensities, and theses four dimensions can be illustrated through individual or combined ion intensity images or as spectra. Many leading edge data analyses have been introduced to IMS data analysis; Principal component analysis degenerates data dimensions and enhances the difference among data.31,32) Hierarchical cluster analysis reveals relationships among molecules and classifies them.31,33) Multivariate curve resolution can reveal mixed spectra from observed single spectrum.34) Markov chain Monte Carlo analysis infers true spectra from incomplete ones, such as in MS/MS.35) Such analyses are helpful for the extraction of the nature of IMS data; however, they are usually too complicated and over engineered. Through extensive research, we found that a simple extension of traditional image processing is the most useful approach.35) Conventional region of interest (ROI) analysis has long been used for the analysis of labeled targets using staining or fluorescent dyes, where localization or distribution of target molecules have been evaluated and ROI-to-ROI are compared. Since mass spectrometry allows for non-targeted studies, IMS enables us to reveal molecules that characterize a specific ROI. Therefore, ROI analysis of IMS data provides an efficient means for screening of biomarkers or key molecules in specific phenomena of interest. Ratio imaging is also popular in conventional image processing, e.g. fluorescence resonance energy transfers (FRET). As already described, IMS can reveal information about huge amount of molecules, which enable to calculate various quantity based on theoretical ideas and mathematical models. An example of such extended ratio analysis is visualization of energy charge, which was introduced to characterize metabolic energy state using number of phosphorylation sites calculated from abundance of ATP, ADP, and AMP.36,37) In the near future, various visualizations will be performed, based on more complex theoretical ideas.
A lot of effort is now being put into finding the best strategies for the extraction of useful information through processes such as baseline correction, denoising and normalization. A strong, collective effort is also put into finding common data formats such as imzML to enable a straightforward sharing of data sets between research groups.38)
Quantification and data normalizationFor quantitation, it is important that the ion signal intensities truly reflect the actual on-tissue concentrations of the analyte in question. One should therefore take into account i) the ionization yield of the respective analytes, ii) matrix deposition homogeneity and iii) the varying tissue- or compartment specific properties that might affect the ionization.
The first applications described for MALDI IMS were developed for protein or peptide analysis, followed by lipid analysis applications. Recently, interest in drug imaging has increased.5,39,40) Imaging of drug distribution is a crucial step in assessing drug function, and currently used methods such as positron emission tomography (PET) or whole body autoradiography (WBA) depend on labeling the drug compound with a radioactive tag. Not only is labeling a costly and time consuming procedure, but also, the subsequent detection process is targeted to the label rather than the drug compound itself, hampering the possibility to find and identify drug metabolites. In contrast, IMS offers the detection of drug molecules as well as their metabolites. Moreover, there is a possibility for un-biased detection of treatment related molecular alterations or reactions in the same tissue sections. Recently, the Andrén group presented a quantification method that is useful for the analysis of a pre-selected target molecule.18) Standard solutions of the target compound are spotted at known concentrations onto control tissue. The spots (5–8 in total) then serve as references for a calibration curve. An analogue to the target, e.g. a deuterated analogue, is added to the matrix solution to correct for ionization biases that the molecule of interest displays. Using the analysis strategy described in Fig. 3, Kallback et al. reached therapeutic levels (pmol) in their detection. Normalizing the data to the deuterated molecule to compensate for local ionization effects generated the best fit, with a correlation coefficient R2=0.985.

Fig. 3. Quantitation: structural overview of the quantitation software design. Figure reprint by permission, Källback et al. J. Proteomics 75(16): 4941–4951, 2012.
A similar approach for the analysis of drug distribution and quantitation in tissue was recently presented by Hamm et al. where they added a “pseudo internal standard,” i.e. the molecule of interest, at known concentrations to the matrix solution and spraying this mix onto control tissue sections. By collecting ion intensity data over a whole body section, drug specific as well as tissue specific ion suppression effects could be analyzed and summarized in a normalization factor denoted the Tissue Extinction Coefficient, TEC. As a consequence, the amount of “drug per area of tissue” could be determined (which in theory should be convertible to “drug per gram of tissue”). Using this method, the drugs propranolol and olanzapine were analyzed at limits of detection (LODs) down to 0.006 pmol/mm2 and 0.3 pmol/mm2, respectively. The IMS data correlated well with results using analytical techniques such as Quantitative Whole Body Autoradiography (QWBA) or LC-MSMS.41)