Abstract
We report the vibrational properties of Li4FeH6 with the highest gravimetric hydrogen density in Fe-based complex hydrides. The Fourier transform infrared (FTIR) spectrum shows a broad and weak peak at 600–1000 cm−1 and an intense one at 1400–1800 cm−1, which are assigned with the aid of first-principles calculations to be the H–Fe–H bending modes, and the antisymmetric Fe–H stretching modes, respectively. From the obtained peak frequency of asymmetric stretching modes of Li4FeH6, the Fe–H bond length is estimated to be 1.6 Å, which is in good agreement with the one predicted by first-principles calculations.
1. Introduction
Iron(Fe)-based complex hydrides, which contain Fe-hydrido complex anion [FeH6]4−, have been the subject of extensive investigation because of practical interest as potential hydrogen storage materials.1–8) In the complex hydrides, [FeH6]4− is generally counterbalanced by alkaline-earth metals (e.g. Mg2FeH6) or mixed alkali/transition metals (e.g. LiYFeH6),3,9) and the gravimetric hydrogen densities reach 5.5 mass% when Mg is adopted as a countercation. Very recently, we have successfully synthesized a novel complex hydride, Li4FeH6, using a high pressure cubic-anvil-type apparatus based on a prediction by first-principles calculations.6,7) This is the first complex hydride of which [FeH6]4− is counterbalanced by alkali metal elements, and has the highest gravimetric hydrogen density of 6.7 mass% in the Fe-based complex hydrides.
The crystal structure of Li4FeH6 was theoretically elucidated to have a trigonal K4CdCl6-type with a = 7.97 Å and c = 9.76 Å in the space group R-3c (No. 167) and Z = 6 (Fig. 1), which is the same structure type as that of the isomorphic Li4RuH6 and Li4OsH6.9) In the complex hydride, Fe is octahedrally coordinated by six hydrogen atoms with Fe–H bond length of 1.61 Å.6) The [FeH6]4− is surrounded with eight cations (Li atoms) which form corner-sharing cube as shown in Fig. 1. This is similar to the edge-sharing cube in the related compounds M′2TH6 (M′ = divalent metal cation), in spite of having different crystal structures (note that M′2TH6 adopts a face-centered cubic K2PtCl6-type structure).10) Although the experimental confirmation of the crystal structure, especially the hydrogen positions, has been strongly desired, it is technically difficult to synthesize a large volume of samples enough for the structure analyses, using the high pressure cubic-anvil-type apparatus.

In this regard, we have recently reported a unique way to estimate the bond length between the center atom and the hydrogen in the related Ru-hydrido complex anion using the vibrational properties. The idea is based on a linear relationship between the frequencies of antisymmetric Ru–H stretching modes and the Ru–H bond length in [RuH6]4−.11) This method would allow for the estimation of the bond length even in the case of a tiny amount of sample obtained.
In this study, we first investigated the vibrational properties of Li4FeH6 in detail (especially, the hydrogen atomic motions), using Fourier transform infrared (FTIR) spectroscopy combined with first-principles calculations. Based on the results, the Fe–H bond length was estimated.
2. Experimental Procedure
2.1 Synthesis
The starting materials, LiH (95% Sigma-Aldrich) and Fe (99.9% Sigma-Aldrich) powders were mixed in a molar ratio of 6:1 and the mixture was pressed into a pellet with a diameter of 1.0 mm and a thickness of 1.0 mm in the same manner as in Ref. 7). The pellet, encapsulated in a pyrolytic boron nitride reaction capsule, was loaded together with a hydrogen source AlH312,13) into a NaCl capsule with a diameter of 1.5 mm and a height of 3.5 mm. The pellet reacted with hydrogen fluid, which was supplied from the hydrogen source, to synthesize Li4FeH6 at 5.5 GPa and 700℃ for 24 hours by using a cubic-anvil-type apparatus. The synthetic conditions were determined based on in-situ synchrotron x-ray diffraction data measured at BL14B1 of SPring-8. The details of the high-temperature and high-pressure synthesis techniques are described in Refs. 7, 14–16).
All samples were handled in an Ar-gas-filled glove box with a dew point below 183 K and with less than 1 ppm of O2 to avoid hydro-oxidation.
2.2 Characterizations
All samples were characterized by X-ray powder diffraction (PANalytical X'PERT PRO diffractometer) using Cu Kα radiation (λ = 1.5406 Å for Kα1 and 1.5444 Å for Kα2).
The vibrational spectra of Li4FeH6 were measured using FTIR (Thermo Scientific Nicolet iN10) spectroscope. For the FTIR spectroscopic studies, a thin sample, approximately a few μm thick, was prepared in an Ar-gas-filled diamond anvil cell, and transmission spectra were measured with a microscope FTIR spectrometer.
2.3 First-principles calculations
The lattice dynamics of 22-atom primitive unit cell of Li4FeH6 were studied using first-principles calculations based on density-functional theory (DFT). We used plane-wave basis sets and the projector augmented-wave method17,18) within the generalized gradient approximation (GGA) of Perdew, Burke, and Ernzerhof,19) as implemented in the Vienna Ab-Initio Simulation Package (VASP).20,21) Well-converged plane-wave basis sets with cutoff energy of 800 eV were employed. The Brillouin-zone sampling was performed using the special k-points method22) with an 8 × 8 × 8 mesh. The structural parameters were taken from Ref. 6). The zone-center phonon frequencies were obtained using first-principles perturbation theory (DFPT).
3. Results and Discussions
3.1 Synthesis
Figure 2 shows the X-ray diffraction patterns of the as-milled and the hydrogenated 6LiH + Fe with a simulated X-ray diffraction pattern of Li4FeH6. Only the peaks from the Fe metal are observed in the pattern of the as-milled sample because of low X-ray scattering from LiH composed of light elements. After the hydrogenation, new peaks are observed and indexed by a trigonal unit cell with a = 7.989(6) Å and c = 9.651(9) Å using indexing programs.23,24) This is in good agreement with previously reported unit cell parameters of Li4FeH6.6,7) Thus, Li4FeH6 was successfully synthesized. Although we attempted to optimize synthesis conditions for a higher quality sample with less amount of Fe by adjusting the starting material ratios, ball-milling times, as well as the synthesis temperature and duration, it was found that the present condition (5.5 GPa and 700℃ for 24 hours on the ball milled 6LiH and Fe) yielded the highest purity of Li4FeH6 in our experiments. Saitoh et al., reported that high temperature at around 900℃ was necessary to obtain a high purity Li4FeH6 in order to accelerate the reaction kinetics.7) Hydrogen pressure higher than 6 GPa pressure was required to avoid subsequent decomposition of Li4FeH6 during synthesis. Although the sample is not a single phase, the quality was good enough for the FTIR to access the hydrogen atomic motion in Li4FeH6.

Figure 3 shows the FTIR spectrum of Li4FeH6. The spectrum has broad and weak peaks at 600–1000 and an intense peak at 1400–1800 cm−1. We performed first-principles phonon calculations to assign the experimentally observed frequencies to the vibrational modes. The calculated zone-center phonon frequencies are summarized in Table 1. Since the primitive unit cell of Li4FeH6 consists of 22 atoms, there are a total of 63 optical phonon modes. The calculated frequencies between 1470 and 1710 cm−1 originate from the Fe–H stretching modes, whereas those ranging from 710 to 950 cm−1 are related to the H–Fe–H bending modes. The remaining frequencies are associated with the Li translational or FeH6 librational modes. Among these frequencies, there are a total of 28 IR active modes, which are shown by tick marks in Fig. 3. From a comparison of the observed and calculated frequencies, the broad and weak peaks at 600–1000 cm−1 and the intense peak at 1400–1800 cm−1 in the experimental FTIR spectrum can be understood to originate, respectively, from the H–Fe–H bending modes, and the antisymmetric Fe–H stretching modes.
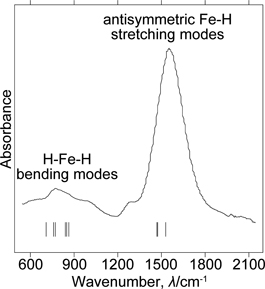
Table 1
Zone-center optical phonon frequencies calculated for the 22 atom primitive cell of Li
4FeH
6. Infrared and Raman activities are shown in parentheses: IR = infrared active; R = Raman active.
| Symmetry |
Mode |
Frequency (cm−1) |
| A1g (R) |
FeH6 librational/
Li translational |
156 |
510 |
|
|
|
|
| A2g |
238 |
416 |
474 |
533 |
|
|
| A1u |
223 |
308 |
|
|
|
|
| A2u (IR) |
232 |
370 |
533 |
|
|
|
| Eg |
170 |
236 |
381 |
415 |
580 |
588 |
| Eu (IR) |
186 |
216 |
245 |
347 |
421 |
|
| A1g (R) |
H–Fe–H bending |
944 |
|
|
|
|
|
| A2g |
924 |
|
|
|
|
|
| A1u |
744 |
891 |
|
|
|
|
| A2u (IR) |
714 |
870 |
|
|
|
|
| Eg (R) |
887 |
914 |
|
|
|
|
| Eu (IR) |
765 |
777 |
846 |
855 |
|
|
| A1g (R) |
Fe–H stretching |
1700 |
|
|
|
|
|
| A2g |
1681 |
|
|
|
|
|
| A1u |
1535 |
|
|
|
|
|
| A2u (IR) |
1474 |
|
|
|
|
|
| Eg (R) |
1688 |
1709 |
|
|
|
|
| Eu (IR) |
1480 |
1535 |
|
|
|
|
The observed FTIR frequencies related to the Fe–H stretching modes of Li4FeH6 are very close to those reported for Ca2FeH6.25) Supposing that the frequencies of the anti-symmetric Fe–H stretching modes are linear with Fe–H bond length as reported for the Ru-based complex hydrides,11) the Fe–H bond length in Li4FeH6 should be comparable to that in Ca2FeH6, and is estimated to be 1.6 Å. This is in good agreement with that predicted by first-principles calculations6).
4. Conclusion
We successfully synthesized Li4FeH6 at 5.5 GPa and 700℃ for 24 hours and investigated the vibrational properties. From the FTIR spectroscopic studies, the antisymmetric Fe–H stretching modes were observed at around 1560 cm−1. Based on a relationship between the frequencies of the anti-symmetric modes and Fe–H bond distances in [FeH6]4−, the Fe–H bond distances was estimated to be 1.6 Å, which was consistent with the previously predicted value by first-principles calculations.
Acknowledgements
The authors would like to thank Ms. H. Ohmiya and Ms. N. Warifune for technical supports, and the use of SR16000 supercomputing resources at the Center for Computational Materials Science of the Institute for Materials Research, Tohoku University. This research was supported by the JSPS KAKENHI Grant Numbers 16K06766, 16H06119, and 25220911 from MEXT, Japan and Collaborative Research Center on Energy Materials in IMR (E-IMR), Tohoku University. The synchrotron radiation experiments were performed at BL14B1 of SPring-8 with the approval of Japan Atomic Energy Agency (JAEA) (Proposal Nos. 2013B3602 and 2013B3614).
REFERENCES
- 1) B. Bogdanović, A. Reiser, K. Schlichte, B. Spliethoff and B. Tesche: J. Alloy. Compd. 345 (2002) 77–89. 10.1016/S0925-8388(02)00308-0
- 2) M. Polanski, T. Płociński, I. Kunce and J. Bystrzycki: Int. J. Hydrogen Energ. 35 (2010) 1257–1266. 10.1016/j.ijhydene.2009.09.010
- 3) M. Matsuo, H. Saitoh, A. Machida, R. Sato, S. Takagi, K. Miwa, T. Watanuki, Y. Katayama, K. Aoki and S. Orimo: RSC Adv. 3 (2013) 1013–1016. 10.1039/C2RA22497F
- 4) K. Miwa, S. Takagi, M. Matsuo and S. Orimo: J. Phys. Chem. C 117 (2013) 8014–8019. 10.1021/jp3122159
- 5) S. Takagi, T.D. Hamphries, K. Miwa and S. Orimo: Appl. Phys. Lett. 104 (2014) 203901. 10.1063/1.4878775
- 6) S. Takagi, T. Ikeshoji, T. Sato, K. Aoki and S. Orimo: J. Jpn. Inst. Met. Mater. 77 (2013) 604–608. 10.2320/jinstmet.JC201310
- 7) H. Saitoh, S. Takagi, M. Matsuo, Y. Iijima, N. Endo, K. Aoki and S. Orimo: APL Mat. 2 (2014) 076103. 10.1063/1.4886219
- 8) S. Takagi and S. Orimo: Scr. Mater. 109 (2015) 1–5. 10.1016/j.scriptamat.2015.07.024
- 9) M. Kritikos, D. Noréus, A.F. Andresen and P. Fischer: J. Solid State Chem. 92 (1991) 514–519. 10.1016/0022-4596(91)90357-N
- 10) K. Yvon: Chimia (Aarau) 52 (1998) 613–619.
- 11) T. Sato, S. Takagi, M. Matsuo, K. Aoki, S. Deledda, B.C. Hauback and S. Orimo: Mater. Trans. 55 (2014) 1117–1121. 10.2320/matertrans.MG201403
- 12) F.M. Brower, N.E. Matzek, P.F. Reigler, H.W. Rinn, C.B. Roberts, D.L. Schmidt, J.A. Snover and K. Terada: J. Am. Chem. Soc. 98 (1976) 2450–2453. 10.1021/ja00425a011
- 13) K. Ikeda, S. Muto, K. Tatsumi, M. Menjo, S. Kato, M. Bielmann, A. Züttel, C.M. Jensen and S. Orimo: Nanotechnology 20 (2009) 204004. 10.1088/0957-4484/20/20/204004
- 14) A. Kamegawa, Y. Goto, R. Kataoka, H. Takamura and M. Okada: Renew. Energy 33 (2008) 221–225. 10.1016/j.renene.2007.05.008
- 15) H. Saitoh, A. Machida and K. Aoki: Chin. Sci. Bull. 59 (2014) 5290–5301. 10.1007/s11434-014-0543-8
- 16) S. Takagi, Y. Iijima, T. Sato, H. Saitoh, K. Ikeda, T. Otomo, K. Miwa, T. Ikeshoji, K. Aoki and S. Orimo: Angew. Chem. Int. Ed. 54 (2015) 5650–5653. 10.1002/anie.201500792
- 17) P.E. Blöchl: Phys. Rev. B 50 (1994) 17953–17979. 10.1103/PhysRevB.50.17953
- 18) G. Kresse and D. Joubert: Phys. Rev. B 59 (1999) 1758–1775. 10.1103/PhysRevB.59.1758
- 19) J.P. Perdew, K. Burke and M. Ernzerhof: Phys. Rev. Lett. 77 (1996) 3865–3868. 10.1103/PhysRevLett.77.3865
- 20) G. Kresse and J. Hafner: Phys. Rev. B 47 (1993) 558–561. 10.1103/PhysRevB.47.558
- 21) G. Kresse and J. Furthmüller: Phys. Rev. B 54 (1996) 11169–11186. 10.1103/PhysRevB.54.11169
- 22) H.J. Monkhorst and J.D. Pack: Phys. Rev. B 13 (1976) 5188–5192. 10.1103/PhysRevB.13.5188
- 23) P.-E. Werner, L. Eriksson and M. Westdahl: J. Appl. Crystallogr. 18 (1985) 367–370. 10.1107/S0021889885010512
- 24) P.-E. Werner: Ark. Kemi 31 (1969) 513–516.
- 25) H. Hagemann, V. D'Anna, L.M. Lawson Daku, S. Gomes, G. Renaudin and K. Yvon: J. Phys. Chem. Solids 72 (2011) 286–289. 10.1016/j.jpcs.2011.01.001