Tunability of Mg2Si Bandgap by Formation of Mg2(Si, C) with an Anti-Fluorite Structure Examined by First-Principles Calculations
2019 Volume 60 Issue 9 Pages 1873-1880

Details
2019 Volume 60 Issue 9 Pages 1873-1880
We used first-principles calculations to investigate the effects of replacing Si atoms in Mg2Si with C atoms to tune its bandgap and enhance its thermoelectric performance. First-principles calculations suggest that the substitution of Si by C atoms in the Mg2Si lattice forms Mg8Si4−zCz (z = 1, 2, and 3) with a sustained anti-fluorite structure, which results in an unexpected bandgap contraction. However, the bandgap of Mg8Si4(1−x)C4x composed of a supercell structure of a rhombohedral Mg2Si primitive cell and Si-substitution with C has a wide bandgap when the C/Si ratio is sufficiently high. The formation enthalpies from Mg, Si, and C (diamond) are negative under pressures greater than ca. 15 GPa.

Fig. 6 Bandgap dependences of Mg8Si4 (□), Mg8C4 (○), Mg8Si3C1 (▲), Mg4Si1C1 (●), and Mg8Si1C3 (×) on their unit-cell volume. Numerical values attached to these symbols indicate the assumed applied pressures in the unit of GPa during the geometrical optimization procedures.
Magnesium silicide (Mg2Si) is a narrow-gap semiconductor, which has attracted interest as a promising and eco-friendly thermoelectric semiconductor in the temperature range of 600–900 K at ambient pressure. Many studies have been performed on Mg2Si-based materials, as reviewed by Gonçalves and Godart1) and Cheng et al.2) However, the thermoelectric performance of these materials is insufficient for practical applications and further improvements are required. The performance of thermoelectric devices depends on a number of physical properties, and particularly on the bandgap between electrons and holes, as theoretically suggested by Sofo and Mahan.3) Therefore, it is of vital importance to tune the bandgap of the materials to optimize device performance for given environmental conditions. It has been claimed that the optimum bandgap of a direct bandgap semiconductor should not be lower than 6kBT and may be higher, depending on the dominant scattering mechanism, where T is the operating temperature of the device and kB is the Boltzmann constant.
To enhance the thermoelectric performance of Mg2Si in the relatively high temperature region, widening of the Mg2Si bandgap is necessary. From this viewpoint, a number of experimental and theoretical studies have been performed to tune the bandgaps of alkaline-earth silicide semiconductors. These include investigations on Mg2Si, BaSi2, and Si-clathrates, which are promising candidate materials for solar energy conversion. Examples include: (1) Mg substituted by isovalent heavier elements, i.e., Ca, Sr, or Ba, in the Mg2Si lattice;4,5) (2) Si substituted by isovalent Ge;6) (3) BaSi2 lattices substituted by isovalent light elements (Ca, Sr);7,8) (4) BaSi2 lattices substituted with alkali metals;9) (5) Si in BaSi2 lattices substituted with isovalent carbon;10) (6) variation of the encapsulated alkali metal M and substitution of the Si atom with a group 13 element in silicon clathrates of the form M8Si38A8 (A = Ga, Al, and In);11) and (7) substitution of Mg in the Mg2Si lattice by transition metals to achieve a hoping chemical pressure effect.12)
In 2014, Balout et al. used a density functional theory (DFT) approach in a theoretical investigation of the effects of tensile and compressive strain on the electronic and thermoelectric properties of Mg2Si.13) They stated that isotropic tensile stress or negative pressure enhanced thermoelectric performance. Substitution of Mg or Si with heavier isovalent elements produces a negative chemical pressure through volume expansion. However, Mg substitution with heavier elements, such as Ca, Sr, and Ba, is not expected to be possible because their half-silicides X2Si (X = Ca, Sr) have the anti-cotunnite structure and the solubility of these elements into the Mg2Si lattice is quite limited.4,5) The solid solubilities of Ge and Sn into Mg2Si are expected to be high because Mg2Ge and Mg2Sn have the same structure as that of Mg2Si. However, their indirect bandgaps are 0.74 and 0.36 eV, respectively, which are narrower than that of Mg2Si (0.77 eV) and a solid solution of Mg2SixGe1−x or Mg2SixSn1−x would have narrower bandgaps than that of Mg2Si.
To realize a negative pressure and increase the bandgap, carbon (C) may have advantages over Ge and Sn as a solid solution element for Mg2Si. Carbon is more electronegative than Si and causes a downward shift of the whole valence band, which widens the bandgap. However, there are two problems to be considered.
First, Mg2C with the anti-fluorite structure was unknown until Kurakevych et al.’s work under high pressure.14) Their successful synthesis of Mg2C with an anti-fluorite structure suggests the possibility of bandgap tuning of Mg2Si by forming a solid solution of Mg2Si and Mg2C with a suitable bandgap for thermoelectric energy conversion. Second, C incorporation might cause a positive chemical pressure effect through volume contraction, which would have the opposite effect to our expectations.
To determine which factors of electronegativity or volume contraction dominate, we performed 1st-principles calculations of Mg8Si4(1−x)C4x (x = 0, 1, 2, 3, and 4) with the anti-fluorite structure. During the calculations, we found that substitution of Si with C atoms in the Mg2Si lattice decreased the bandgap to lower values than even that of Mg2Si. To better understand the discontinuity between the result for Mg8Si1C3 and that for Mg8C4, which has been confirmed to have a much wider bandgap than that of Mg8Si4, we extended the scope of this work from our original assumption to include compounds with C/Si ratios higher than 3 for Mg8Si1C3.
Mg2Si has an anti-fluorite structure, belonging to the space group No. 225. In the Mg2Si unit cell, Mg atoms occupy 8c sites [Wyckoff notation] at the eight (±1/4, ±1/4, ±1/4) positions, whereas Si atoms are located at the cube corners and the three face centers, (4a sites). Therefore, the unit cell contains four formula units, corresponding to the stoichiometry of Mg8Si4. However, it can be reduced to a primitive rhombohedral Mg2Si cell if we adopt a set of primitive vectors pointing from a corner site to any of the three kinds of face-centered points of the cubic lattice, which is composed of Si atoms. The Mg2C considered here also has the same structure as that of Mg2Si.
When some of the Si atoms in the Mg8Si4 cell are substituted with C atoms, the symmetry of the system is lowered from No. 225 to No. 221 when one or three of the Si atoms are substituted, respectively, and to No. 123 when two of the Si atoms are substituted. In the former case, the cells of Mg8Si3C1 and Mg8Si1C3 cannot be reduced further. In the latter case, the cell of Mg8Si2C2 can be reduced to the cell of Mg4Si1C1 with a tetragonal symmetry. First, we performed calculations for Mg8Si4 and Mg8C4 with the assumed symmetry of space group 225, Mg8Si3C1 and Mg8Si1C3 with the assumed symmetry of the space group 221, and Mg4Si1C1 with the assumed symmetry of the space group 123. The calculated structures are summarized in Table 1. We performed several preliminary full-optimization calculations without assuming the space groups of the compounds; however, there were no meaningful differences in these results.

Initially, we considered that sufficient results for bandgap widening of Mg2Si by substitution with C can be obtained by considering the above structures. However, as stated in the introduction, the range of the calculations was not sufficient by itself. We extended the range of the calculation to include compounds with higher C/Si ratios. Details of the construction method of the model compounds will be described later.
2.2 Calculation methodThe calculation method was similar to that used in our previous study of the Ca2Si–Mg2Si system.4) Namely, calculations were performed using the CASTEP code developed by Payne et al.;15) this is a first-principles method based on DFT, with a pseudopotential description of the electron–core interactions and a plane-wave expansion of the wavefunctions. The ultrasoft pseudopotential generated by the Vanderbilt’s scheme16) was used, in which the Mg 2p state was explicitly treated as part of the valence. The Perdew–Wang generalized gradient approximation (GGA)17) were used to approximate the DFT exchange–correlation term.
As for calculation method used here, it should be noted that typical local density approximation (LDA) (and GGA) functionals are known to face so-called ‘bandgap problem’. Several devises have been made to solve this problem such as (A), the so-called LDA+U method18) of including an on-site Coulomb correlation to the effective Hamiltonian of the LDA, (B), hybrid exchange-correlation functional usually constructed as a linear combination of the Hartree–Fock exact exchange functional and any number of exchange-correlation explicit density functionals19,20) and (C), the Tran–Blaha modified Becke-Johnson (mBJ) method.21) Among them, (A) is effective to improve the bandgap of localized systems such as d-semiconductors and insulators but encounters many difficulties in describing the properties of systems with more delocalized electrons such as metals.22) From this fact, it seems that its applicability to s- and p-semiconductors such as Mg2Si is not good. Since (B) and (C) are likely to be promising in reducing the bandgap problem, the applicability of those methods to the present work is kept for future studies. In the present study, we used somewhat classical approximation method, expecting that qualitative guidance would be given by the present calculations and keeping in mind to ensure consistency with our previous studies on Mg2Si and related materials.4,5,8–12,23–26)
Self-consistent iteration convergence in the present study was assumed when the total energy difference between successive cycles was less than 0.001 eV per atom. The maximum error in the calculated energies was therefore approximately ±0.01 eV for Mg8Si4 and similar compounds. The kinetic cutoff energy for the plane-wave expansion (Ecutoff) was set to be 380 eV. To calculate the electronic energies, the Monkhorst–Pack (MP) k-point sampling scheme27) of reciprocal space was used with a spacing of 0.5 nm−1. We performed geometric optimization with the total energy minimization algorithm under the Broyden–Fletcher–Goldfarb–Shannon optimization procedure. The band structures (BSs) along several high-symmetry lines in the Brillouin zone were calculated for the optimized structures. The top of the valence bands was selected to be the zero energy level in drawing the BS diagrams.
As stated above, our initial target is substituted structures of Mg8Si4 with the anti-fluorite structure and the composition of Mg8Si4(1−x)C4x. Therefore, we first describe the results of the terminal compounds of Mg2Si [or, Mg8Si4 (x = 0)] and Mg2C [or, Mg8C4 (x = 1)].
The calculated BS of the optimized Mg2Si under atmospheric pressure (0 GPa) is shown in Fig. 1(a). Plotted numerical values in this and succeeding band structure diagrams are the numbers which express the order, counted from the bottom of the valence band. Because the Mg2p states are explicitly treated as a part of the valence band in the pseudopotential used, 24 bands composed of Mg2p atomic orbitals are present for Mg8Si4 and the other compounds calculated here; however, these bands are omitted from the numbering in this and succeeding figures for simplicity.
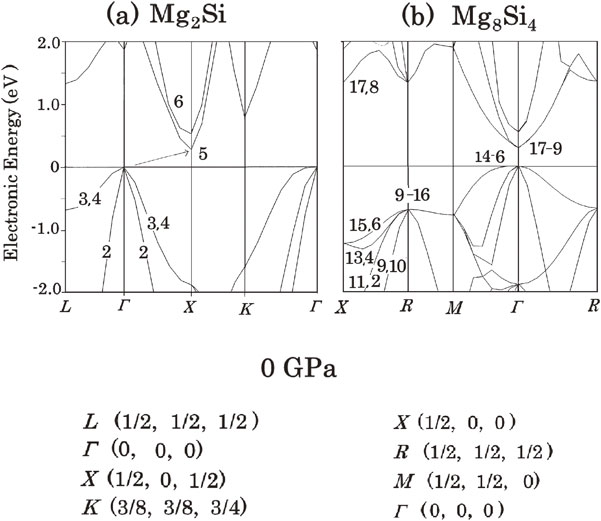
Band structure (BS) of Mg2Si at 0 GPa; (a) Calculated BS for the rhombohedral primitive cell of Mg2Si. (b) Calculated BS for the cubic cell of Mg8Si4. Plotted numerical values in these and succeeding band structure diagrams are the numbers that express the order, counted from the bottom of the valence band.
Mg2Si is an indirect semiconductor where the top of the valence band is located at the Γ point in reciprocal space whereas the bottom of the conduction band is located at the X point. The gap width between them was calculated to be 0.277 eV, which is approximately 36% of the observed value (0.77 eV), owing to the well-known tendency of DFT calculations to underestimate bandgap values.
The calculated BS of the optimized Mg8Si4 under atmospheric pressure is shown in Fig. 1(b). This diagram suggests that Mg8Si4 is a direct semiconductor with a Γ → Γ transition. However, this result is attributed to the fact that both Γ (0, 0, 0) and X (1/2, 0, 1/2) of the reciprocal space of Mg2Si correspond to Γ (0, 0, 0) of the reciprocal space of Mg8Si4. Thus, the indirectness of Mg2Si from Γ to X becomes unclear when the cells of Mg8Si4 are used for band-structure calculations. Hereafter, we decided to consider only the change of the bandgap values of the system and not the directness of the bandgap.
The same can be said for Mg2C. Figure 2(a) shows the calculated BS of Mg2C with an anti-fluorite structure based on a rhombohedral primitive cell, whereas Fig. 2(b) shows that of Mg8C4 based on a cubic unit cell. Here, the calculation was performed for the optimized structure under an applied pressure of 15 GPa because Mg2C is unstable under atmospheric pressure.

Band structure (BS) of Mg2C; (a) Calculated BS for the rhombohedral primitive cell of Mg2C. (b) Calculated BS for the cubic cell of Mg8C4.
Again, Mg2C is an indirect semiconductor with a calculated gap value of 0.874 eV, although it appeared to be a direct semiconductor if we use a cubic unit cell of Mg8C4. We could not compare the calculated bandgap with the observed one because no experimental values have been reported. However, our calculated value is within the range of previous calculations (0.67–0.97 eV).28)
The BSs of Mg2Si and Mg2C changed with applied pressure. Figure 3 shows the calculated BSs of Mg2Si at 30 GPa, calculated based on the rhombohedral primitive cell of Mg2Si, (a), and the cubic cell of Mg8Si4, (b), respectively. The top of the valence bands in Fig. 3(a), the 4th-band, is located at the Γ-point and the bottom of the conduction bands, the 5-th band, is located at the X-point; however, their electronic energy values are reversed in these two; the latter is 0.173 eV lower than the former. This is the so-called state of a ‘negative bandgap’ (or an inverted band structure). This situation is represented in Fig. 3(b) for a cubic unit cell. This energy inversion was calculated to occur at approximately 15 GPa.
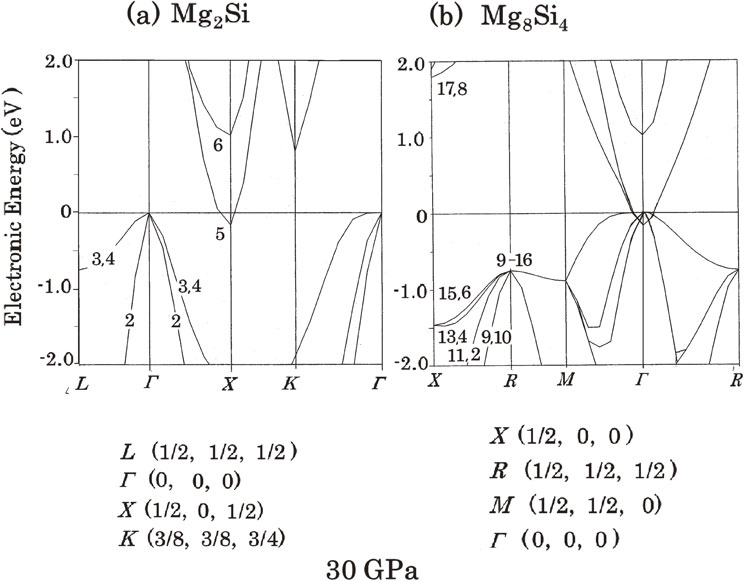
Band structure (BS) of Mg2Si at 30 GPa. (a) Calculated BS for the rhombohedral primitive cell of Mg2Si. (b) Calculated BS for the cubic cell of Mg8Si4.
As for Mg2C, the changes of the bandgap under applied pressures were moderate, but slightly increased from 0.852 eV at 0 GPa to 0.880 eV at 30 GPa, as shown in Fig. 4.

Variation of band structure (BS) of Mg8C4 from 0 to 30 GPa.
Next, we calculated the band structures of Mg8Si4(1−x)C4x with the anti-fluorite structures where Si atoms are partially substituted with C atoms. The calculated BSs of the optimized structures of Mg8Si3C1, Mg8Si1C3, and Mg4Si1C1 at ambient pressure (0 GPa) are shown in Fig. 5(a), (b), and (c), respectively, where the former two belong to the space group 221 whereas the latter belongs to the space group 123.

Band structure of Mg8Si3C1 (a); Mg8Si1C3 (b); and Mg4Si1C1 (c); with the anti-fluorite structures where one to three Si atoms in the Mg8Si4 lattice are substituted with C atoms.
Both Mg8Si3C1 and Mg8Si1C3 are zero-gap semiconductors where the 16-th band (i.e., the highest valence band) and the 17-th band (i.e., the lowest conduction band) are in contact at the only Γ points of their own reciprocal lattice [Fig. 5(a) and (b)]. Conversely, the energy range of the highest valence band, the 8-th band, and the lowest conduction band, the 9-th band, of Mg4Si1C1 overlapped, which indicates that Mg4Si1C1 has a negative energy gap. The result that Si-substitution with C atoms causes a bandgap contraction of Mg2Si is contrary to our presumptions prior to calculations.
One possible reason for this result is that Si substitution with C causes a volumetric contraction of Mg2Si, which decreases the bandgap value. Thus, the volume effect caused by substitution might exceed the chemical effect caused by the difference of the electronegativity between C and Si. To investigate whether predicted bandgap contraction can be explained by the volumetric contraction caused by Si substitution of Mg2Si with C, we decided to investigate the effects of the volume change on the bandgap values of these compounds.
The bandgap dependences of those compounds on their unit-cell volumes are shown in Fig. 6, along with those of Mg2Si and Mg2C, as stated in the previous section.

Bandgap dependences of Mg8Si4 (□), Mg8C4 (○), Mg8Si3C1 (▲), Mg4Si1C1 (●), and Mg8Si1C3 (×) on their unit-cell volume. Numerical values attached to these symbols indicate the assumed applied pressures in the unit of GPa during the geometrical optimization procedures.
As described in the previous section, the bandgap of Mg8Si4 (Mg2Si) decreased with decreasing cell-volume (or as the applied pressure increased from 0 to 30 GPa) in the assumed pressure range for calculations. Conversely, the pressure dependence of the bandgap of Mg8C4 (Mg2C) was small, although the bandgap value slightly increased with decreasing cell-volume (increasing applied pressure).
Calculations of Mg8Si4(1−x)C4x where Si atoms in the Mg8Si4 lattice were partially substituted with C atoms, revealed the following. First, the pressure dependence of the gap values become more moderate than that of Mg8Si4. The bandgap of Mg8Si3C1 has a maximum value of 0 eV at 0 GPa, and decreases at both positive and negative pressures. This result suggests that the bandgap value would not generally have a monotonous dependence on the applied pressure but a maximum value at a certain pressure; however, this was not confirmed within the range of calculated pressures in the present calculations.
Next, the gaps have zero or negative values and do not necessarily increase with changes to the cell volume. This result conflicts with our presumption that Si substitution by C atoms would increase the gap value.
Third, the bandgap value of Mg8Si1C3, marked by the symbol (×), was persistent with the variation of cell-volume. This result suggests that the bandgap contraction was not only caused by the volumetric effect but also by the Brillouin zone folding owing to the superlattice formation by the ordered substitution of atoms. It has been predicted that the band structure of nitrogen (N)-doped graphene changes markedly from a gapless state to states with a definite gap value, depending on the calculated cell-size of the assumed supercell size (or, the concentration of doped N atoms).29) The observed persistency of the contact of the top of the valence band and the bottom of the conduction band at only the Γ point suggested a limiting current in a one-dimensional situation, so-called Luttinger-liquid behavior.
To conclude the present section, our calculations suggest that the bandgap of Mg2Si cannot be widened by substituting Si atoms with C atoms without altering the atomic arrangement. A region of the bandgap = 0.3–0.8 eV in Fig. 6 is blank. Therefore, there is a need for compounds to fill this space. To search for possible compounds with wider bandgaps than that of Mg2Si but narrower than that of Mg2C, we decided to relax our constraints and search for possible compounds with similar atomic arrangements to Mg2Si. We considered, as an example, a structure in which the primitive units of Mg2Si were the same and only the stacking order was different.
In the following section, we describe the BSs of those compounds where all but one Si atom in the superlattice of Mg2Si were substituted with carbon.
3.3 Mg8Si4(1−x)C4x composed of a superlattice of the rhombohedral Mg2Si where all but one Si atom are substituted with C atomsWe constructed 2 × 2 × 1, 2 × 2 × 2, and 3 × 2 × 2 superlattices of the rhombohedral primitive cell of Mg2Si and all but one of the Si atoms were replaced by carbon atoms. The cell compositions thus prepared were Mg8Si1C3, Mg16Si1C7, and Mg24Si1C11, respectively. Those compounds are found to belong to the space groups 65, 225, and 12, respectively. The unit cells, which express their symmetry more directly are Mg16Si2C6, Mg64Si4C28, and Mg48Si2C22, and can be reduced to Mg8Si1C3, Mg16Si1C7, and Mg24Si1C11, respectively, by selecting the proper primitive vectors. The crystallographic data of the former representation, including the fractional coordinates of each atom before geometrical optimization, are shown in Table 2. In the last column of Table 2, the values of the Wyckoff position, not regulated, are described for convenience. These values are generated by the operation of 2 × 2 × 1, 2 × 2 × 2, or 3 × 2 × 2 supercell formation of Mg2Si but slightly varied during the optimization procedure.

The calculated BSs of these compounds at 0 GPa are shown in Fig. 7. In Fig. 7(a), a negative bandgap is found in the BS of Mg8Si1C3, which is different from that of Mg8Si1C3 belonging to the space group No. 221, as previously stated. From an energetic viewpoint, the structure belonging to the Space Group No. 65 was slightly more negative (∼0.16 eV per formula unit) than the structure belonging to the Space Group No. 221; however, further calculations for this structure have not been conducted because the predicted negative bandgap was beyond the scope of the present study.

Band structures of Mg8Si1C3, Mg16Si1C7, and Mg24Si1C11 at 0 GPa.
As shown in Figs. 7(b) and (c), a wider bandgap than that of Mg8Si4 can be expected for Mg16SiC7 and Mg24Si1C11. Thus, the bandgaps of Mg8Si4(1−x)C4x were larger than that of Mg2Si and smaller than that of Mg2C when a high value of x/(1 − x) (0 ≤ x ≤ 1) can be attained. In fact, volume (or, applied pressure) dependences of the bandgap of those compounds, as shown in Fig. 8, indicate that the blank region of the bandgap range in Fig. 6 can be completed with those compounds and bandgap tuning may be possible by varying the composition and applied pressure during the synthesis procedure.

The bandgap dependences of Mg16Si1C7 (■) and Mg24Si1C11 (△) on their unit-cell volume. Numerical values attached to these symbols have the same meaning with those in Fig. 6. Those of Mg8Si4 and Mg8C4 are also showing by □ and ○, respectively, for ease of understanding.
Calculated standard formation enthalpies of those compounds are shown in Fig. 9. Here, the reference states of Mg, Si, and C are metallic Mg with a hexagonal closed-packed structure, semiconducting Si with a diamond structure, and diamond for carbon. The interlayer attraction energy of graphite is hard to evaluate by DFT calculations; hence, graphite, which is an equilibrium phase under ambient conditions, was not selected as the reference state of carbon.

Calculated formation enthalpies of Mg8Si4, Mg16Si1C7, Mg24Si1C11, and Mg8C4.
As shown in Fig. 9, Mg8Si4 with an anti-fluorite structure is more stable than a mixture of Mg, Si, and C throughout the calculated pressure range. However, Mg16Si1C7, Mg24Si1C11, and Mg8C4 are stable at pressures greater than ca. 15 GPa. The results for Mg8C4 agreed well previous calculations by Kurakevych et al.14)
Thus, the bandgaps of the compounds in which the basic structural units of Mg2Si are stacked and all but one of the other Si atoms are substituted with C atoms are wider than that of Mg2Si, if the C/Si ratio is greater than 7, and narrower than that of Mg2C, as expected. These are stable at a high pressure of approximately 15 GPa or more and bandgap tuning is possible through formation of a Mg2Si–Mg2C solid solution. Notably, further investigations are necessary to determine the following possibilities:
In summary, we have calculated the band structures of Mg8Si3C1, Mg8Si1C3, and Mg4Si1C1 with anti-fluorite structures where one to three Si atoms in the Mg8Si4 lattice are substituted with C atoms and Mg8Si4(1−x)C4x composed of a stacked structure (or, superlattice) of the rhombohedral Mg2Si primitive cell where all but one of the Si atoms are substituted with C atoms. Though the possibility of underestimation of the calculated bandgaps derived from the GGA calculation must be reserved, we found that:
The authors would like to express their sincere gratitude to Dr. Naomi Hirayama, the Institute for Solid State Physics, the University of Tokyo, for her informative suggestions on the progress of the DFT calculations.