
- |<
- <
- 1
- >
- >|
-
Tetsu Tsubogo, Saki Aoyama, Rika Takeda, Hiromi Uchiro2018 年 66 巻 9 号 p. 843-846
発行日: 2018/09/01
公開日: 2018/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録The complete synthesis of D-α-tocopherol was achieved using our developed-Ullmann C–O coupling reaction as a key reaction. The synthesis of the core structure of D-α-tocopherol, which is a chiral chromane, has never been reported using intramolecular Ullmann C–O coupling reactions owing to the low reactivity of electron-rich iodoarenes with tertiary alcohols. Because the developed intramolecular C–O coupling reactions prefer electron-rich iodoarenes with tertiary alcohols, we successfully synthesized the chiral chromane core and achieved the total synthesis of D-α-tocopherol.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (542K) HTML形式で全画面表示 -
Yasuhiro Yamashita, Joseph Alexander Macor, Seiya Fushimi, Tetsu Tsubo ...2018 年 66 巻 9 号 p. 847-850
発行日: 2018/09/01
公開日: 2018/09/01
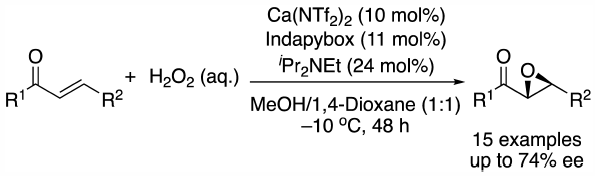 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Asymmetric epoxidation reactions of chalcone derivatives catalyzed by chiral calcium complexes using hydrogen peroxide were developed. The epoxidation reactions proceeded smoothly to afford the desired products in good yields with good enantioselectivities. This is the first example of chiral calcium-catalyzed asymmetric epoxidation reactions using hydrogen peroxide as the terminal oxidant.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (459K) HTML形式で全画面表示
-
Takamasa Suzuki, Yuma Sakisako, Yui Kurihara, Tomohiro Aoki, Toshihiro ...2018 年 66 巻 9 号 p. 851-858
発行日: 2018/09/01
公開日: 2018/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLExcess stripping of stratum corneum (SC) layers by patch-peeling from the skin surface is one cause of skin irritation. High SC hydration by patch occlusion may also cause skin irritation, although the occlusive technique is preferable to increase the skin permeation of topically applied drugs. In the present study, film having a honeycomb structure was selected as the backing layer of a drug-in-adhesive (DIA) patch to reduce peeling of the SC without losing adhesion force to the skin surface, as well as decreasing the skin permeation of a model drug, tulobuterol. The usefulness of the DIA patch with honeycomb film was evaluated by transepidermal water loss (TEWL) changes, amount of SC removed by patch-peeling, distribution pattern of removed SC on the adhesive layer, and water permeation through the patch. Furthermore, skin permeation and release profiles of tulobuterol from the DIA patch were investigated. Significantly (p<0.05) less TEWL change was observed after removal of the patch with a honeycomb film compared with the conventional pressure-sensitive adhesive patch, and no difference in tulobuterol permeation through skin from the patches was confirmed regardless of the type of backing layer. In addition, a lower amount of SC was removed by the peeling of the patch with a honeycomb film. The results suggest that DIA patches with a honeycomb film as a backing layer may be used to achieve less SC removal without reducing the skin permeation of drugs.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (5077K) HTML形式で全画面表示 -
Porntip Benjasirimongkol, Keisuke Ueda, Kenjirou Higashi, Pornsak Sria ...2018 年 66 巻 9 号 p. 859-865
発行日: 2018/09/01
公開日: 2018/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLWe examined the effect of hot–melt extrusion condition on the physical stability of the solid dispersion prepared using partially hydrolyzed polyvinyl alcohol (PVOH). The hot–melt extrusion of indomethacin (IMC) and PVOH mixed at the weight ratio of 3 : 7, 5 : 5 and 7 : 3 was performed either at 170 or 190°C to prepare the IMC/PVOH hot–melt extrudate (HME). Differential scanning calorimetry represented that IMC was mixed with PVOH on a scale of several tens of nanometer in all the HMEs with different weight ratio. 13C solid-state NMR measurement revealed that an intermolecular interaction was formed between a carboxylic group of IMC and a hydroxy group of PVOH in the HMEs. The intermolecular interaction in the HMEs was stronger at the higher extrusion temperature. At the low IMC loading, the IMC molecules could be mixed with the amorphous PVOH at the molecular level, and the remained PVOH without interaction formed the crystal phase. On the other hand, at the high IMC loading, most PVOH could be amorphized by the interaction with IMC, and the excess IMC which did not interact with PVOH formed the IMC-rich domain. The IMC/PVOH HME at the weight ratio of 7 : 3 extruded at higher extrusion temperature showed higher physical stability of amorphous IMC compared with that extruded at lower extrusion temperature. The hot–melt extrusion process at higher temperature provided the rapid melting of PVOH crystal phase, resulted in the homogeneous mixing with IMC and the formation of stronger intermolecular interaction.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1541K) HTML形式で全画面表示 -
 Shinsuke Inuki, Takashi Miyagawa, Shinya Oishi, Hiroaki Ohno2018 年 66 巻 9 号 p. 866-872
Shinsuke Inuki, Takashi Miyagawa, Shinya Oishi, Hiroaki Ohno2018 年 66 巻 9 号 p. 866-872
発行日: 2018/09/01
公開日: 2018/09/01
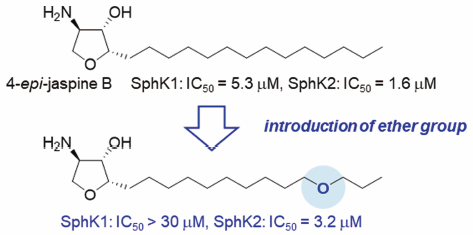 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Sphingosine kinases (SphKs) are key enzymes that regulate sphingosine 1-phosphate production levels, and are involved in a range of cellular processes. Focusing on a hydrophilic residue in the hydrophobic binding pocket of SphKs, we designed and synthesized 4-epi-jaspine B derivatives containing a polar functional group in the lipid tail. A biological evaluation revealed that the introduction of ether groups to the lipid tail of 4-epi-jaspine B modulated its isoform selectivity toward SphKs.
Graphical Abstract Fullsize Image抄録全体を表示Editor's pick4-epi-Jaspine B derivatives containing a polar functional group in the lipid tail were designed and synthesized for the development of sphingosine kinase (SphK) inhibitors, which would be applicable to the treatment of autoimmune and inflammatory disorders. A biological evaluation revealed that the replacement of one methylene at the lipid tail with ether oxygen affected the inhibitory activity of 4-epi-jaspine B, leading to the identification of a selective SphK2 inhibitor.
PDF形式でダウンロード (1219K) HTML形式で全画面表示 -
Daisuke Yamamoto, Hiromasa Ansai, Junichi Hoshino, Kazuishi Makino2018 年 66 巻 9 号 p. 873-879
発行日: 2018/09/01
公開日: 2018/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録A novel aerobic manganese-catalyzed oxophosphorylation reaction of carbon–carbon double bonds of styrene derivatives and vinyl ethers using diethyl H-phosphonates was developed. This direct transformation of alkenes to β-ketophosphonate readily proceeded at room temperature via the direct incorporation of molecular oxygen present in air (open flask).
Graphical Abstract Fullsize Image抄録全体を表示Editor's pickA novel aerobic manganese-catalyzed oxophoshporylation reaction of carbon–carbon double bonds of styrene derivatives and vinyl ethers using diethyl H-phosphonates was developed. This direct transformation of alkenes to b-ketophosphonates readily proceeded at room temperature via the incorporation of molecular oxygen present in air (open flask). All of the experimental procedures in this reaction can be performed in air without cooling, heating, or high pressure. This methodology serves an alternative approach to produce β-ketophosphonates because of its simplicity and mildness.
PDF形式でダウンロード (797K) HTML形式で全画面表示 -
Ryu Yamasaki, Yutaka Honjo, Ai Ito, Kazuo Fukuda, Iwao Okamoto2018 年 66 巻 9 号 p. 880-884
発行日: 2018/09/01
公開日: 2018/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録We have discovered a spontaneous reaction of N,O-diaryl carbamates to afford symmetrical N,N′-diarylureas. Optimization of the conditions indicated that N,N-dimethylformamide (DMF) was the best solvent and triethylamine (Et3N) was the best additive for this transformation. The reaction requires the presence of aryl groups on the nitrogen and oxygen atoms of carbamates. Substrates bearing an electron-donating methoxy group on either of the aryl groups reacted slowly under these conditions.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (472K) HTML形式で全画面表示
-
Luo-Na Wang, Hao-Wen Jiang, Jin-Long Li, Jing-Ya Li2018 年 66 巻 9 号 p. 885-886
発行日: 2018/09/01
公開日: 2018/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Type 2 diabetes is characterized by hyperglycemia derived from insulin resistance in periphery tissue. Effects of skeletal muscle on glucose disposal are closely related to insulin resistance. The potential effects on mitochondrial function of loesenerine, a macrocyclic spermidine alkaloid from the aerial part of Euonymus fortunei (TURCZ.) HAND.-MAZZ were observed after a high-throughout screening based on mitochondrial membrane potential (MMP) assay. Further pharmacological studies revealed that loesenerine activates AMP-activated protein kinase (AMPK) pathway through increasing ADP/ATP ratio by inhibiting mitochondrial respiration. In addition, loesenerine induced 1.07-, 1.14-, and 1.22-fold increment of glucose uptake in C2C12 cells at the concentrations of 20, 40 and 80 µmol/L, respectively. Meanwhile, incubated with loesenerine for 12 h increased glucose consumption in a dose-dependent manner in C2C12 cells. This is the first report that macrocyclic spermidine alkaloid possesses potential hypoglycemic activity in vitro.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (521K) HTML形式で全画面表示 -
Miho C. Emoto, Kota Sasaki, Koya Maeda, Hirotada G. Fujii, Shingo Sato2018 年 66 巻 9 号 p. 887-891
発行日: 2018/09/01
公開日: 2018/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録The drug-nitroxide radical hybrid-compound 7-N-((2,2,5,5-tetramethylpyrrolidine-1-yloxy(PROXYL))-3-yl-methyl)theophylline (3) was synthesized by coupling 7-N-tosyltheophylline with 3-hydroxymethyl-PROXYL, HMP). The stability of 3 relative to that of HMP was examined in the presence of the anti-oxidant, ascorbic acid (AsA). The initial reduction rate constants of 3 and HMP were 11.9±5.3 and 6.1±5.2 M−1 min−1, respectively. In the presence of glutathione (GSH), these constants increased slightly to 22.3±6.8 and 9.1±2.4 M−1 min−1, respectively. Two-dimensional cranial electron paramagnetic resonance imaging of mice intravenously injected with 3 via the tail vein revealed that probe 3 enters the mouse brain by passing through the blood–brain barrier (BBB).
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1258K) HTML形式で全画面表示 -
Akihito Kiguchiya, Reiko Teraoka, Toshiyasu Sakane2018 年 66 巻 9 号 p. 892-895
発行日: 2018/09/01
公開日: 2018/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLEvaluating the stability of drugs over ranges of various environmental conditions is necessary to ensure the quality of the drug throughout the shelf life. In this study, we used indomethacin, which is well known as a photosensitive drug, and evaluated the photostability of prepared crystals. HPLC analysis revealed that all the indomethacin crystals were degraded by light exposure. However, the indomethacin crystals involving solvates had different degrees of photodegradation and different solid-state UV/Vis spectra. The value of the absorption integral in the UVA range related closely to the photodegradation of indomethacin crystals involving solvates. Therefore, it is easy to compare the photosensitivity among the crystals without actual analytical data, by use of a suitable analytical method and using light exposure samples. Moreover, it is possible to predict the value of photodegradation ratio from the solid-state UV/Vis spectra of indomethacin crystals. Therefore, this method may provide key information for selecting the most appropriate crystal form of photosensitive drugs.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (572K) HTML形式で全画面表示 -
Siriporn Phutthatiraphap, Yoshihiro Hayashi, Takuto Fujii, Atsushi Kos ...2018 年 66 巻 9 号 p. 896-900
発行日: 2018/09/01
公開日: 2018/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLTo investigate the inhibitory effect of a commercial proton pump inhibitor (lansoprazole) on the gastric proton pump H+,K+-ATPase in vitro, we used orally disintegrating (OD) tablets including original brand-name and generic tablets. In the course of the development of generic products, dissolution and clinical tests are necessary to ensure their bioequivalence to the original brand-name products; by contrast, there is almost no opportunity to demonstrate their activity in vitro. This study initially compared the similarity of the dissolution of test generic tablets with that of the original brand-name tablets. The dissolution tests for 15 and 30-mg lansoprazole tablets found their dissolution properties were similar. Subsequently, the dissolution media were sampled and then their effects on the H+,K+-ATPase activity were measured using tubulovesicles prepared from the gastric mucosa of hogs. We confirmed that the inhibitory effects of the generic tablets on H+,K+-ATPase activity were consistent with those of the original brand-name tablets. Furthermore, lansoprazole contents in each tablet estimated from their inhibitory effects were in good agreement with their active pharmaceutical ingredient content. To our knowledge, this is the first technical report to compare the in vitro biochemical activity of lansoprazole OD tablets between the original brand-name and generic commercial products.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (478K) HTML形式で全画面表示 -
Hongshan Yu, Yu Wang, Chunying Liu, Jiamei Yang, Longquan Xu, Guanheng ...2018 年 66 巻 9 号 p. 901-906
発行日: 2018/09/01
公開日: 2018/09/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Ginsenoside Rb1 is an important saponin of ginseng(s); however, Rb1, with 3-O- and 20-O-sugar moieties, has low bioavailability. Here, we report the derivatization of ginsenoside Rb1 to completely generate six types of highly bioactive minor ginsenoside Rg3 and its derivatives by FeCl3 catalysis, the reaction conditions are similar to enzymatic reaction conditions. In FeCl3 catalysis, the only 20-O-sugar-moiety of ginsenoside Rb1 was decomposed into the minor ginsenosides Rk1 and Rg5 with newly produced C-20 ethylene bands; but also hydrolyzed into 20(S)-Rg3 and 20(R)-Rg3; subsequently the C-24(25) ethylene bands of 20(S)-Rg3 and 20(R)-Rg3 were hydrated to 20(S)-25-OH-Rg3 and 20(R)-25-OH-Rg3. After separation of reaction mixture from 34 g ginsenoside-Rb1 by silica-gel-column, the 3.3 g sample I of TLC top-band consisting of Rg5 and Rk1, 8.7 g sample II of TLC middle-band consisting of 20(S)-Rg3 and 20(R)-Rg3, 3.5 g sample III of TLC bottom-band consisting of unknown product-I and -II including 20(S)-25-OH-Rg3, were obtained. The sample III consisting of unknown product-I and -II was purified by crystallization, and identified to 20(S)-25-OH-Rg3 and 20(R)-25-OH-Rg3 by HPLC-Evaporative Light Scattering Detector (ELSD) and NMR. Therefore, six types of minor-ginsenosides Rk1, Rg5, 20(S)-Rg3, 20(R)-Rg3, 20(S)-25-OH-Rg3 and 20(R)-25-OH-Rg3 were successfully prepared from ginsenoside Rb1 by FeCl3 catalysis. FeCl3 has low toxicity and is inexpensive, and the reaction conditions are similar to enzymatic reaction conditions; thus, this method is applicable to the development of ginseng-based drugs.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (907K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|