
- |<
- <
- 1
- >
- >|
-
Koyo Nishida2020Volume 68Issue 7 Pages 559
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
JOURNAL FREE ACCESS FULL-TEXT HTMLDownload PDF (180K) Full view HTML
-
 Hidemasa Katsumi, Shugo Yamashita, Masaki Morishita, Akira Yamamoto2020Volume 68Issue 7 Pages 560-566
Hidemasa Katsumi, Shugo Yamashita, Masaki Morishita, Akira Yamamoto2020Volume 68Issue 7 Pages 560-566
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLBone metastases can cause high morbidity and mortality, often developing as they advance, especially in patients with prostate and breast cancers. Most drugs are rarely distributed to the bone and are hence pharmacologically ineffective in treating bone metastases. The development of drug targeting technologies is required for the efficient treatment of bone metastases. To date, numerous bone-targeting ligands, including tetracyclines, bisphosphonates, aspartic acid, and aptamers have been developed and used for bone-targeted delivery of anti-tumor drugs, peptide/protein drugs, nucleic acid drugs, and diagnostic imaging agents. The conjugates of drugs with bone-targeting ligands were first developed in the field of bone drug targeting systems; macromolecular carriers and nanoparticles modified with these bone-targeting ligands have also been developed. Additionally, antibodies to prostate-specific membrane antigen (PSMA) and human epidermal growth factor receptor 2 (HER2) are used in active targeting bone metastatic prostate cancer and breast cancer, respectively. Some conjugates using antibodies to PSMA and HER2 were developed and used in clinical trials. In this review, recent challenges in the development of bone-targeted delivery systems and strategies for the treatment of bone metastasis have been summarized. Future development of novel drug formulations in order to optimize targeted drug delivery in the treatment of bone metastasis have also been discussed.
Graphical Abstract Fullsize ImageView full abstractEditor's pickThis paper analyzes and summarizes the development of novel drug formulations in order to optimize targeted drug delivery in the treatment of bone metastasis using bone-targeting ligands and antibodies to metastatic cancer. Metastases of the bone is a common development especially in patients with breast and prostate cancers. However, most drugs are inefficiently distributed to the bone and are hence pharmacologically suboptimal in treating metastases in the bone. This paper could be useful for the development of drug targeting technologies for the treatment of bone metastasis.
Download PDF (1623K) Full view HTML -
Koki Ogawa, Naoya Kato, Shigeru Kawakami2020Volume 68Issue 7 Pages 567-582
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLBecause the brain is the most important human organ, many brain disorders can cause severe symptoms. For example, glioma, one type of brain tumor, is progressive and lethal, while neurodegenerative diseases cause severe disability. Nevertheless, medical treatment for brain diseases remains unsatisfactory, and therefore innovative therapies are desired. However, the development of therapies to treat some cerebral diseases is difficult because the blood–brain barrier (BBB) or blood–brain tumor barrier prevents drugs from entering the brain. Hence, drug delivery system (DDS) strategies are required to deliver therapeutic agents to the brain. Recently, brain-targeted DDS have been developed, which increases the quality of therapy for cerebral disorders. This review gives an overview of recent brain-targeting DDS strategies. First, it describes strategies to cross the BBB. This includes BBB-crossing ligand modification or temporal BBB permeabilization. Strategies to avoid the BBB using local administration are also summarized. Intrabrain drug distribution is a crucial factor that directly determines the therapeutic effect, and thus it is important to evaluate drug distribution using optimal methods. We introduce some methods for evaluating drug distribution in the brain. Finally, applications of brain-targeted DDS for the treatment of brain tumors, Alzheimer’s disease, Parkinson’s disease, and stroke are explained.
Graphical Abstract Fullsize ImageView full abstractEditor's pickBecause brain disorders such as glioma, Alzheimer’s disease and Parkinson’s disease are lethal, or cause severe symptoms, medical treatment is needed. Recently, with the progress of understanding pathology, new therapeutic candidates have been developed. However, there are still unmet medical needs in the medication of brain disorder. This is mainly because most drugs cannot cross blood-brain barrier (BBB). To solve this problem, drug delivery system (DDS) using some approaches (e.g. antibody, nanocarrier, ultrasound irradiation) is being studied for overcoming BBB. In this review, we comprehensively reviewed on the development and therapeutic application of brain-targeted DDS.
Download PDF (1847K) Full view HTML -
Yu Ishima, Toru Maruyama, Masaki Otagiri, Tatsuhiro Ishida2020Volume 68Issue 7 Pages 583-588
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLA unique phenomenon in solid tumors, the enhanced permeability and retention (EPR) effect is now well known in the development of macromolecular anticancer therapy. However, cancers with low vascular permeability have posed a challenge for these EPR based therapeutic systems. An intrinsic vascular modulator, such as nitric oxide (NO), could augment the endogenous EPR effect. However, the most important aim has been to construct an effective NO delivery system for cancer. Since it is well known that human serum albumin is one of the most important endogenous NO transport proteins in human circulation, for more than a decade we have demonstrated that S-nitrosated human serum albumin dimer (SNO-HSA-Dimer) becomes an enhancer of the EPR effect. Here, we summarize the enhanced effect of SNO-HSA-Dimer on the anticancer effect of macromolecular anticancer drugs such as PEGylated liposomal doxorubicin (Doxil®). In C26-bearing mice with highly permeable vasculature, SNO-HSA-Dimer is able to increase more 3-fold the tumor accumulation of these anticancer drugs, thereby tripling their anticancer effects. Interestingly, the tumor accumulation of Doxil® in B16-bearing mice, which are characterized by a low permeable vasculature, increased more than 6-fold in the presence of SNO-HSA-Dimer, and the improved accumulation of Doxil® led to both increased survival and decreased tumor volume. These results strongly suggest that the more cancer is refractory, the more the SNO-HSA-Dimer could enhance the EPR effect via an endogenous albumin transport (EAT) system. Accordingly, we conclude that the EAT system is promising as a drug delivery system (DDS) strategy for refractory cancer therapy.
Graphical Abstract Fullsize ImageView full abstractEditor's pickPassive targeting can be applied to a relatively wide range of cancer types due to the enhanced permeability and retention (EPR) effect that is the basis of its theory. However, recent clinical data indicate that the tumor accumulation is only approximately 10% of the dose, and satisfactory results are most rarely obtained using only EPR effect strategy. In this article, the authors introduce the strategy of enhancing the EPR effect using S-nitrosated human serum albumin dimer (SNO-HSA Dimer) and the DDS strategy utilizing the endogenous albumin transport (EAT) system of tumor cells and its future development.
Download PDF (2448K) Full view HTML -
Tomoyuki Okuda, Hirokazu Okamoto2020Volume 68Issue 7 Pages 589-602
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
 JOURNAL FREE ACCESS FULL-TEXT HTML
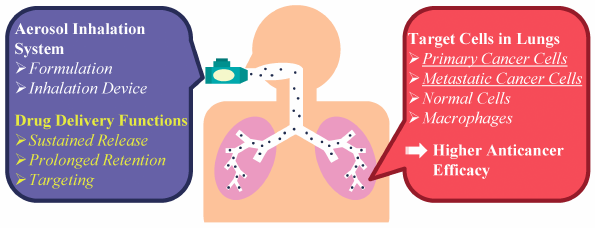
JOURNAL FREE ACCESS FULL-TEXT HTMLInhaled lung cancer therapy is promising because of direct and noninvasive drug delivery to the lungs with low potential for severe systemic toxicity. Thus chemotherapeutic drugs have been administered clinically by nebulization of solution or suspension formulations, which demonstrated their limited pulmonary absorption and relatively mild systemic toxicity. In all these clinical trials, however, there was no obviously superior anticancer efficacy in lung cancer patients even at the maximum doses of drugs limited by pulmonary toxicity. Therefore methods that deliver both higher anticancer efficacy and lower pulmonary toxicity are strongly desired. In addition to the worldwide availability of pressured metered dose inhalers (pMDIs) and dry powder inhalers (DPIs) to treat local respiratory diseases, recent innovations in medicines and technologies are encouraging next steps toward effective inhaled lung cancer therapy with new therapeutic or drug delivery concepts. These include the discovery of target cells/molecules and drug candidates for novel cancer therapy, the development of high-performance inhalation devices for effective pulmonary drug delivery, and the establishment of manufacturing technologies for functional nanoparticles/microparticles. This review highlights the present situation and future progress of inhaled drugs for lung cancer therapy, including an overview of available inhalation devices, pharmacokinetics, and outcomes in clinical trials so far and some novel formulation strategies based on drug delivery systems to achieve enhanced anticancer efficacy and attenuated pulmonary toxicity.
Graphical Abstract Fullsize ImageView full abstractEditor's pickInhaled lung cancer therapy is promising because of direct and noninvasive drug delivery to the lungs with low potential for severe systemic toxicity. In all clinical trials with nebulization of chemotherapeutic drugs, however, there was no obviously superior anticancer efficacy in lung cancer patients even at the maximum doses of drugs limited by pulmonary toxicity. Thus the addition of further drug delivery functions including sustained release, prolonged retention, and targeting in the lungs has been strongly desired to achieve enhanced anticancer efficacy and attenuated pulmonary toxicity of inhaled chemotherapeutic drugs and other drug candidates.
Download PDF (668K) Full view HTML -
Shintaro Fumoto, Koyo Nishida2020Volume 68Issue 7 Pages 603-612
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLCancer treatments have improved significantly during the last decade but are not yet satisfactory. Combination therapy is often administered to improve efficacy and safety. Drug delivery systems can also improve efficacy and safety. To control the spatiotemporal distribution of drugs, nanotechnology involving liposomes, solid lipid nanoparticles, and polymeric micelles has been developed. Co-delivery systems of multiple drugs are a promising approach to combat cancer. Synergistic effects and reduced side effects are expected from the use of co-delivery systems. In this review, we summarize various co-delivery systems for multiple drugs, including small-molecule drugs, nucleic acids, genes, and proteins. Co-delivery of drugs with different properties is relatively difficult, but some researchers have succeeded in developing such co-delivery systems. Environment-responsive carrier designs can control the release of cargos. Although their preparation is more complicated than that of mono-delivery systems, co-delivery systems can simplify clinical procedures and improve patient QOL.
Graphical Abstract Fullsize ImageView full abstractEditor's pickCombination therapies using multiple drugs are the effective strategies to treat tumors. Various therapeutic regimens have been developed. Here, co-delivery of multiple drugs to tumor tissues using nanotechnology is theoretically useful to improve the efficacy and safety of the regimens. In this review, the authors summarized the current state of co-delivery systems of multiple drugs, including small-molecular chemotherapeutic drugs, proteins, nucleic acids and gene medicines. Especially, they pointed out the importance of selection of the combination, targeted delivery of multiple drugs, and controlled release of drugs based on environment-responsive mechanisms. Co-delivery systems are the promising approach even for immunotherapy using checkpoint inhibitors.
Download PDF (667K) Full view HTML
-
Yefang Zou, Zhuoxian Cao, Jie Wang, Xiaoxue Chen, Yan-qin Chen, Yan Li ...2020Volume 68Issue 7 Pages 613-617
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
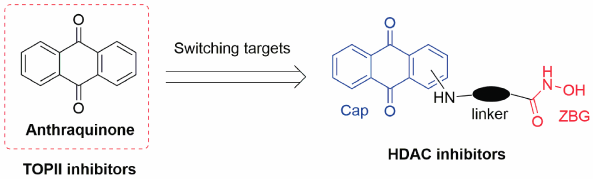
Supplementary materialAlthough anthraquinone derivatives possess significant antitumor activity, most of them also displayed those side effects like cardiotoxicity, mainly owing to their inhibition of topoisomerase II of DNA repair mechanisms. Our raised design strategy by switching therapeutic target from topoisomerase II to histone deacetylase (HDAC) has been applied to the design of anthraquinone derivatives in current study. Consequently, a series of novel HDAC inhibitors with a tricylic diketone of anthraquinone as a cap group have been synthesized. After screening and evaluation, compounds 4b, 4d, 7b and 7d have displayed the comparable inhibition in enzymatic activity and cell proliferation than that of Vorinostat (SAHA). Notably, compound 4b showed certain selectivity of antiproliferative effects on cancer cell lines over non-cancer cell lines.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (856K) Full view HTML
-
Dan Wang, Yue Meng, Xuelei Wang, Guimin Xia, Qiang Zhang2020Volume 68Issue 7 Pages 618-627
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
Advance online publication: May 12, 2020 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialA novel polymer (PEG2000-carborane), self-assembling into spherical vesicles (boron-containing vesicles, BCVs), could be quickly taken up by tumor cells and had an enhance stability in the bloodstream in previous study. To have more comprehensive understanding of BCVs, endocytic mechanism and cytotoxicity assessment were conducted. The results showed that BCVs were taken up in the intact form with cholesterol-dependent pathway during endocytosis, and BCVs exhibited nearly no cytotoxicity. BCVs could accumulate within tumors for at least 24 h. The data would provide reference information and guidance for BCVs’ multifunctional application serving as a boron delivery agent for boron neutron capture therapy (BNCT), a hydrophilic and/or hydrophobic drug carrier and a diagnostic imaging fluorescent probe.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (3351K) Full view HTML -
Jianfeng Pan, Chenfang Miao, Yuanting Chen, Jiahui Ye, Zhenzhen Wang, ...2020Volume 68Issue 7 Pages 628-634
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
Advance online publication: April 24, 2020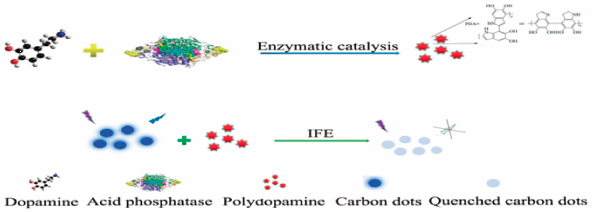 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialFacile and effective detection of dopamine (DA) plays a significant role in current clinical applications. Substantially, special optical nanomaterials are important for fabricating easy-to-control, cheap, selective, and portable fluorescence DA sensors with superior performance. Herein, carbon dots (CDs) prepared from melting method were applied as signal to establish a simple but effective fluorescence strategy for DA determination based on the enzymatic activity of acid phosphatase (ACP), which induces DA to form polydopamine (pDA). The formed pDA caused by the enzymatic oxidization of ACP toward DA can interact with CDs through the inner filter effect. Such behavior effectively quenched the CDs’ fluorescence. The degree of fluorescence quenching of CDs was positively correlated with the DA content. Under the optimized reaction conditions, the proposed fluorescence method exhibited a comparable analytical performance with other DA sensors with good selectivity. Furthermore, this method has been successfully applied to detect DA in DA hydrochloride injection and human serum samples. It shows that this method features potential practical application value and is expected to be used in clinical research.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (2488K) Full view HTML -
Shoichi Kuroda, Yohei Kobashi, Madoka Kawamura, Kenichi Kawabe, Fumiya ...2020Volume 68Issue 7 Pages 635-652
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLSodium-glucose cotransporter 1 (SGLT1) is the primary transporter for glucose absorption from the gastrointestinal tract. While C-phenyl D-glucitol derivative SGL5213 has been reported to be a potent intestinal SGLT1 inhibitor for use in the treatment of type 2 diabetes, no SGLT1 selectivity was found in vitro (IC50 29 nM for hSGLT1 and 20 nM for hSGLT2). In this study we found a new method of synthesizing key intermediates 12 by a one-pot three-component condensation reaction and discovered C-phenyl D-glucitol 41j (TP0454614), which has >40-fold SGLT1 selectivity in vitro (IC50 26 nM for hSGLT1 and 1101 nM for hSGLT2). The results of our study have provided new insights into the structure–activity relationships (SARs) of the SGLT1 selectivity of C-glucitol derivatives.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (1274K) Full view HTML -
Kyosuke Tanaka, Hiroyuki Kobayashi, Sayaka Suzuki, Satoshi Shibuya, Hi ...2020Volume 68Issue 7 Pages 653-663
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialThe discovery of a novel class of state-dependent voltage-gated sodium channel (NaV)1.7 inhibitors is described. By the modification of amide or urethane bond in NaV1.7 blocker III, structure–activity relationship studies that led to the identification of novel NaV1.7 inhibitor 2i (DS01171986) were performed. Compound 2i exhibited state-dependent inhibition of NaV1.7 without NaV1.1, NaV1.5 or human ether-a-go-go related gene (hERG) liabilities at concentrations up to 100 μM. Further biological profiling successfully revealed that 2i possessed potent analgesic properties in a murine model of neuropathic pain (ED50: 3.4 mg/kg) with an excellent central nervous system (CNS) safety margin (> 600 fold).
Graphical Abstract Fullsize ImageView full abstractDownload PDF (1274K) Full view HTML -
Hiroyuki Yoshida, Yasuhiro Abe, Naomi Tomita, Ken-ichi Izutsu2020Volume 68Issue 7 Pages 664-670
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialResearch from the past decade has shown that the buffer capacities of intestinal fluids are much lower than those in the media used for dissolution test of many solid formulations. The purpose of this study was to elucidate the effect of buffer capacity on the dissolution profiles of highly soluble drug products, using metoclopramide (a biopharmaceutics classification system [BCS] class III drug) tablets as a model. The dissolution profiles of three metoclopramide products were obtained in Japanese pharmacopeia dissolution medium (pH 1.2 and 6.8), diluted medium with low buffer capacity comparable to that of gastrointestinal fluid, and other biorelevant media. One product showed slower dissolution in the medium with lower buffer capacity (bio-relevant, diluted compendial solution), but substantially similar dissolution in the compendial test solutions. Disintegration difference was implied to be involved in the different dissolution profiles depending on the medium buffer capacity. This study indicated the importance of media buffer capacity as a factor inducing different dissolution between products of highly soluble active pharmaceutical ingredients. The diluted compendial media would be a useful alternative to biorelevant media for the detection of the different formulation performances depending on the buffer capacities.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (1695K) Full view HTML
-
Sirada Boonyaketgoson, Yongle Du, Ana L. Valenciano Murillo, Maria B. ...2020Volume 68Issue 7 Pages 671-674
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialChromatographic separation of the acetone extracts from the twigs and barks of Artocarpus lakoocha led to the isolation of the one new flavanone, lakoochanone (1), together with eleven known compounds (2–12). Lakoochanone (1) and moracin C (4) exhibited weak antiplasmodial activity against Plasmodium falciparum Dd2 with IC50 values of 36.7 and 33.9 µM, respectively. Moreover, moracin C (4) and sanggenofuran B (5) showed cytotoxic activity against A2780 cell line with the respective IC50 values of 15.0 and 57.1 µM. In addition, cyclocommunin (7) displayed strong antimycobacterial activity against Mycobacterium tuberculosis H37Ra with the minimum inhibitory concentration (MIC) value of 12.3 µM.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (402K) Full view HTML -
Naoto Kojima, Hiromi Hayashi, Hiroki Iwasaki, Masayuki Yamashita2020Volume 68Issue 7 Pages 675-678
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialThe details of the total syntheses of C2′-fluorinated analogs of solamin, an antitumor annonaceous acetogenin, are described. Fluorine was enantioselectively introduced at the C2′-position by organocatalytic α-fluorination of the aldehyde according to a previously reported method. C2′-fluorinated solamin and its C2′-diastereomer were synthesized by the Sonogashira coupling of a tetrahydrofuran fragment and fluorine-containing γ-lactone fragments.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (485K) Full view HTML -
Yuki Murata, Hitomi Iwasa, Mio Matsumura, Shuji Yasuike2020Volume 68Issue 7 Pages 679-681
Published: July 01, 2020
Released on J-STAGE: July 01, 2020
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
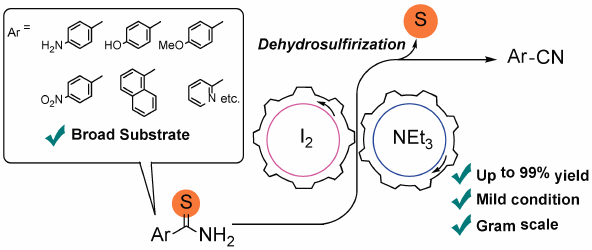
Supplementary materialA simple general method for the synthesis of nitriles using the inexpensive and easy to handle iodine (I2) is described herein. The reaction of thioamides with I2 in the presence of triethylamine at room temperature under aerobic conditions afforded various nitriles bearing aryl, vinyl, and alkyl groups in good-to-excellent yields. This method was also effective for conversion from thioureas to cyanamides.
Graphical Abstract Fullsize ImageView full abstractDownload PDF (397K) Full view HTML
- |<
- <
- 1
- >
- >|