
Volume 18, Issue 1
Special Issue: "Element Strategy for Catalysts and Batteries: Approach from Theoretical and Computational Chemistry"
Displaying 1-12 of 12 articles from this issue
- |<
- <
- 1
- >
- >|
Foreword
-
2019 Volume 18 Issue 1 Pages A1-A2
Published: 2019
Released on J-STAGE: February 20, 2019
Download PDF (360K) Full view HTML
Accounts
-
2019 Volume 18 Issue 1 Pages 1-8
Published: 2019
Released on J-STAGE: February 20, 2019
Advance online publication: December 31, 2018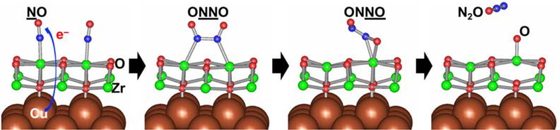 Download PDF (3240K) Full view HTML
Download PDF (3240K) Full view HTML -
2019 Volume 18 Issue 1 Pages 9-17
Published: 2019
Released on J-STAGE: February 20, 2019
Advance online publication: February 07, 2019 Download PDF (1497K) Full view HTML
Download PDF (1497K) Full view HTML -
2019 Volume 18 Issue 1 Pages 18-28
Published: 2019
Released on J-STAGE: February 20, 2019
Advance online publication: February 05, 2019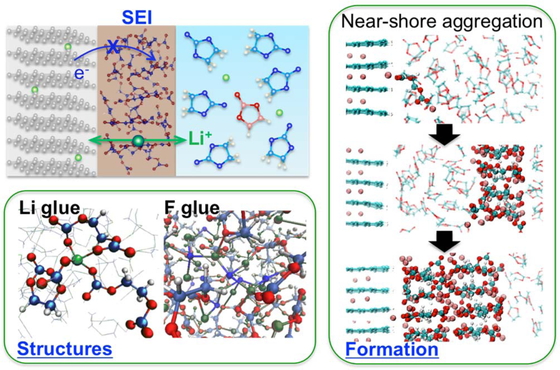 Download PDF (2823K) Full view HTML
Download PDF (2823K) Full view HTML -
2019 Volume 18 Issue 1 Pages 29-37
Published: 2019
Released on J-STAGE: February 20, 2019
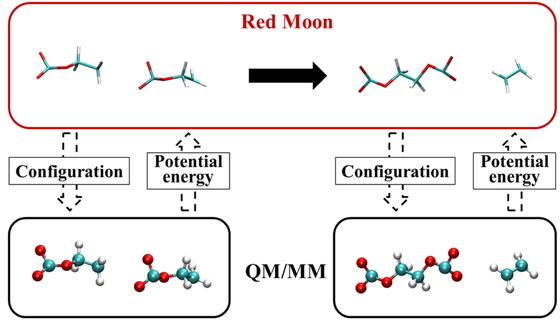 Download PDF (3529K) Full view HTML
Download PDF (3529K) Full view HTML
Reviews
-
2019 Volume 18 Issue 1 Pages 38-48
Published: 2019
Released on J-STAGE: February 20, 2019
Advance online publication: January 12, 2019 Download PDF (3128K) Full view HTML
Download PDF (3128K) Full view HTML -
2019 Volume 18 Issue 1 Pages 49-63
Published: 2019
Released on J-STAGE: February 20, 2019
Advance online publication: February 14, 2019 Download PDF (5346K) Full view HTML
Download PDF (5346K) Full view HTML
General Papers
-
2019 Volume 18 Issue 1 Pages 64-69
Published: 2019
Released on J-STAGE: February 20, 2019
Advance online publication: February 07, 2019 Download PDF (1850K) Full view HTML
Download PDF (1850K) Full view HTML -
2019 Volume 18 Issue 1 Pages 70-77
Published: 2019
Released on J-STAGE: February 20, 2019
Advance online publication: January 12, 2019 Download PDF (1712K) Full view HTML
Download PDF (1712K) Full view HTML -
2019 Volume 18 Issue 1 Pages 78-83
Published: 2019
Released on J-STAGE: February 20, 2019
Advance online publication: February 09, 2019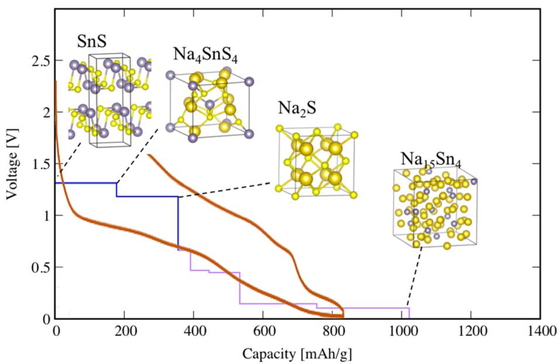 Download PDF (1814K) Full view HTML
Download PDF (1814K) Full view HTML -
2019 Volume 18 Issue 1 Pages 84-94
Published: 2019
Released on J-STAGE: February 20, 2019
Advance online publication: February 07, 2019 Download PDF (3243K) Full view HTML
Download PDF (3243K) Full view HTML -
2019 Volume 18 Issue 1 Pages 95-101
Published: 2019
Released on J-STAGE: February 20, 2019
Advance online publication: December 31, 2018 Download PDF (1056K) Full view HTML
Download PDF (1056K) Full view HTML
- |<
- <
- 1
- >
- >|