All issues

Volume 56 (2013)
- Issue 6 Pages 357-
- Issue 5 Pages 253-
- Issue 4 Pages 183-
- Issue 3 Pages 109-
- Issue 2 Pages 61-
- Issue 1 Pages 1-
Volume 56, Issue 2
Displaying 1-7 of 7 articles from this issue
- |<
- <
- 1
- >
- >|
Review Paper
-
Toshimasa Takanohashi, Shinya Sato, Ryuzo TanakaArticle type: Review Paper
2013 Volume 56 Issue 2 Pages 61-68
Published: 2013
Released on J-STAGE: May 01, 2013
 JOURNAL FREE ACCESSThe repression of coke precursor formation is key to heavy oil upgrading. Asphaltenes are known to form aggregates that may be responsible for coke precursor formation and the consequent deactivation of catalysts. A Supra-Molecular Asphaltene Relaxation Technology (SMART) is introduced herein and may be applied to asphaltene aggregates in order to reduce their detrimental effects. The nature of asphaltene aggregates was examined using molecular simulation methods. Molecular mechanics and molecular dynamics calculations on asphaltenes obtained from vacuum residues revealed that the most stable conformations of asphaltene aggregates were those held together by several noncovalent interactions. At 673 K, where decomposition reactions begin, aggregates formed of aromatic-aromatic stacking interactions were still stable. These stable aggregates likely comprised heavier oil fractions such as coke precursors. Changes induced in these aggregated structures by pretreatment with various solvents were investigated. Some stacking interactions could be disrupted in quinoline at 573 K but remained stable when the aggregates were pretreated in 1-methylnaphthalene. Autoclave experiments showed that the coke yield after pyrolysis at 713 K was significantly decreased when the asphaltene was presoaked in quinoline for 1 h. In contrast, pretreatment with 1-methylnaphthalene resulted in negligible changes in coke yield. The results of both simulations and autoclave experiments suggest that, when aggregates were presoaked in quinoline, some aromatic stacking interactions were disrupted and molecular mobility increased. This prevented the asphaltenes from polymerizing via condensation reactions between aromatic rings. Thus, coke yields after pretreatment in quinoline were relatively low. The contribution of each type of interaction (aromatic stacking, aliphatic ring entanglement, heteroatom interactions, and hydrogen bonding) to the overall aggregation energy of asphaltene was estimated using an imaginary simulation technique. Aromatic-aromatic interactions accounted for approximately 50 % of the total aggregate interactions. Contributions from aliphatic side-chain entanglement and heteroatom interactions were around 27 % and 20 %, respectively. The combination of these interactions stabilized the asphaltene aggregates.View full abstractDownload PDF (925K)
JOURNAL FREE ACCESSThe repression of coke precursor formation is key to heavy oil upgrading. Asphaltenes are known to form aggregates that may be responsible for coke precursor formation and the consequent deactivation of catalysts. A Supra-Molecular Asphaltene Relaxation Technology (SMART) is introduced herein and may be applied to asphaltene aggregates in order to reduce their detrimental effects. The nature of asphaltene aggregates was examined using molecular simulation methods. Molecular mechanics and molecular dynamics calculations on asphaltenes obtained from vacuum residues revealed that the most stable conformations of asphaltene aggregates were those held together by several noncovalent interactions. At 673 K, where decomposition reactions begin, aggregates formed of aromatic-aromatic stacking interactions were still stable. These stable aggregates likely comprised heavier oil fractions such as coke precursors. Changes induced in these aggregated structures by pretreatment with various solvents were investigated. Some stacking interactions could be disrupted in quinoline at 573 K but remained stable when the aggregates were pretreated in 1-methylnaphthalene. Autoclave experiments showed that the coke yield after pyrolysis at 713 K was significantly decreased when the asphaltene was presoaked in quinoline for 1 h. In contrast, pretreatment with 1-methylnaphthalene resulted in negligible changes in coke yield. The results of both simulations and autoclave experiments suggest that, when aggregates were presoaked in quinoline, some aromatic stacking interactions were disrupted and molecular mobility increased. This prevented the asphaltenes from polymerizing via condensation reactions between aromatic rings. Thus, coke yields after pretreatment in quinoline were relatively low. The contribution of each type of interaction (aromatic stacking, aliphatic ring entanglement, heteroatom interactions, and hydrogen bonding) to the overall aggregation energy of asphaltene was estimated using an imaginary simulation technique. Aromatic-aromatic interactions accounted for approximately 50 % of the total aggregate interactions. Contributions from aliphatic side-chain entanglement and heteroatom interactions were around 27 % and 20 %, respectively. The combination of these interactions stabilized the asphaltene aggregates.View full abstractDownload PDF (925K) -
Kenji Wada, Hiroki Miura, Saburo Hosokawa, Masashi InoueArticle type: Review Paper
2013 Volume 56 Issue 2 Pages 69-79
Published: 2013
Released on J-STAGE: May 01, 2013
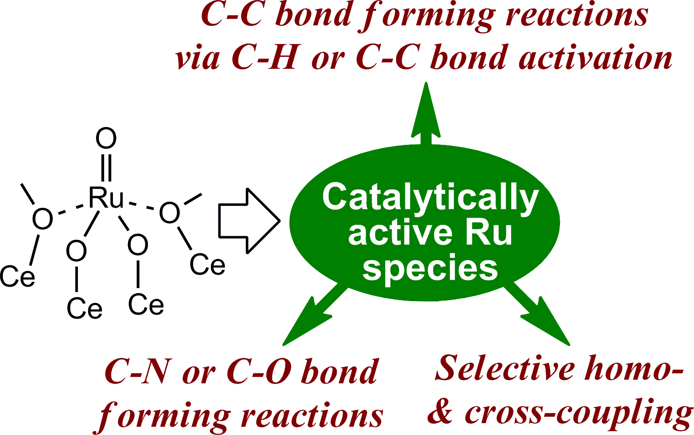 JOURNAL FREE ACCESSSolid Ru/CeO2 catalysts, which are effective for the catalysis of various highly atom-efficient organic transformations such as dehydrogenative synthesis of indole, regio- and stereo-selective coupling of unsaturated hydrocarbons, C–C bond-forming reactions via the activation of aromatic C–H bonds, and selective addition of carboxylic acids to alkynes, have been developed. For these reactions, CeO2-supported and, in some cases, ZrO2-supported catalysts show excellent activities, whereas ruthenium catalysts on other oxide supports are not effective. Spectroscopic studies have revealed that various types of catalytically active, low-valent ruthenium species are generated from ruthenium(IV) oxo species that are exclusively formed on the surface of CeO2 under appropriate conditions. These solid ruthenium catalysts can be recycled without loss of activity or significant leaching of ruthenium species into the solution, and these features, as well as the wide range of applicable reactions, make the present catalytic system quite attractive from both practical and environmental perspectives.View full abstractDownload PDF (1496K)
JOURNAL FREE ACCESSSolid Ru/CeO2 catalysts, which are effective for the catalysis of various highly atom-efficient organic transformations such as dehydrogenative synthesis of indole, regio- and stereo-selective coupling of unsaturated hydrocarbons, C–C bond-forming reactions via the activation of aromatic C–H bonds, and selective addition of carboxylic acids to alkynes, have been developed. For these reactions, CeO2-supported and, in some cases, ZrO2-supported catalysts show excellent activities, whereas ruthenium catalysts on other oxide supports are not effective. Spectroscopic studies have revealed that various types of catalytically active, low-valent ruthenium species are generated from ruthenium(IV) oxo species that are exclusively formed on the surface of CeO2 under appropriate conditions. These solid ruthenium catalysts can be recycled without loss of activity or significant leaching of ruthenium species into the solution, and these features, as well as the wide range of applicable reactions, make the present catalytic system quite attractive from both practical and environmental perspectives.View full abstractDownload PDF (1496K)
Regular Paper
-
Masatoshi Nagai, Hiroyuki Tominaga, Shigetaka KaiArticle type: Regular Paper
2013 Volume 56 Issue 2 Pages 80-87
Published: 2013
Released on J-STAGE: May 01, 2013
 JOURNAL FREE ACCESSThe distribution of active sites on a nitrided CoMo/Al2O3 catalyst was studied based on a non-parametric determination method using the conversion rate of hydrodesulfurization of dibenzothiophene. This method makes use of a Fredholm integral equation of the first kind and provides the activity distribution of the catalytic active sites. These active sites were discussed based on the reaction products during the reaction, the surface properties by CO adsorption and X-ray photoelectron spectroscopy. The distribution function revealed two active sites of the nitrided catalyst with different rate constants for the hydrogenation of dibenzothiophene on Site I and for the C–S hydrogenolysis on Site II. Site II had a 58.3 times higher rate and 1.27 times the population than Site I for the 973 K-nitrided catalyst.View full abstractDownload PDF (403K)
JOURNAL FREE ACCESSThe distribution of active sites on a nitrided CoMo/Al2O3 catalyst was studied based on a non-parametric determination method using the conversion rate of hydrodesulfurization of dibenzothiophene. This method makes use of a Fredholm integral equation of the first kind and provides the activity distribution of the catalytic active sites. These active sites were discussed based on the reaction products during the reaction, the surface properties by CO adsorption and X-ray photoelectron spectroscopy. The distribution function revealed two active sites of the nitrided catalyst with different rate constants for the hydrogenation of dibenzothiophene on Site I and for the C–S hydrogenolysis on Site II. Site II had a 58.3 times higher rate and 1.27 times the population than Site I for the 973 K-nitrided catalyst.View full abstractDownload PDF (403K) -
Hideo Nagata, Shun-ichi Hirayama, Yuki Johno, Nobuyuki FurukawaArticle type: Regular Paper
2013 Volume 56 Issue 2 Pages 88-93
Published: 2013
Released on J-STAGE: May 01, 2013
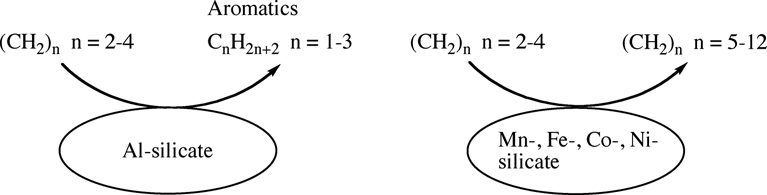 JOURNAL FREE ACCESSMetallosilicates containing manganese or iron group metals were prepared using a rapid crystallization method. The acidities of these metallosilicates and catalytic activities for the conversion of light olefins to gasoline fraction were investigated. The acidity and catalytic activity of the synthesized Mn–silicate did not depend on the manganese content. In contrast, the acid strengths and catalytic activities of metallosilicates containing iron group metals (iron-group-silicates) depended on the content and type of iron group metal. The acidity of the iron-group-silicates increased in the following order:
JOURNAL FREE ACCESSMetallosilicates containing manganese or iron group metals were prepared using a rapid crystallization method. The acidities of these metallosilicates and catalytic activities for the conversion of light olefins to gasoline fraction were investigated. The acidity and catalytic activity of the synthesized Mn–silicate did not depend on the manganese content. In contrast, the acid strengths and catalytic activities of metallosilicates containing iron group metals (iron-group-silicates) depended on the content and type of iron group metal. The acidity of the iron-group-silicates increased in the following order:
Co–silicate < Fe–silicate < Ni–silicate.
The selectivity for light paraffins and aromatic hydrocarbons over iron-group-silicates in conversion of light olefins increased in the same order. Consequently, Co–silicate had the lowest selectivity for aromatic hydrocarbons among all the iron-group-silicates for the conversion of light olefins into gasoline fraction.View full abstractDownload PDF (319K)
Research Note
-
Yasuharu Kanda, Toru Araki, Masatoshi Sugioka, Yoshio UemichiArticle type: Research Note
2013 Volume 56 Issue 2 Pages 94-101
Published: 2013
Released on J-STAGE: May 01, 2013
 JOURNAL FREE ACCESSCatalytic properties of ruthenium phosphide (Ru2P) supported on silica for hydrodenitrogenation (HDN) of pyrrole were compared with those of Ru/SiO2 catalyst to clarify the effect of phosphidation. At higher W/F (652 g h mol−1), pyrrole conversion over Ru/SiO2 catalyst remarkably decreased with increasing reduction temperature. In contrast, pyrrole conversion over P-added Ru (Ru–P/SiO2) catalyst at the same W/F was enhanced with increasing reduction temperature due to Ru2P formation. This activity was higher than that of sulfided NiMoP/Al2O3 catalyst but lower than that of Ru/SiO2 catalyst at the same W/F. At lower W/F (130-391 g h mol−1), Ru–P/SiO2 catalyst showed higher activity and stability for pyrrole HDN than Ru/SiO2 catalyst. The cracking products (almost all CH4) were formed over Ru/SiO2 catalysts and butanes were formed over Ru–P/SiO2 catalysts. The results of CO adsorption and TEM images revealed that the particle size of Ru–P/SiO2 catalyst was smaller than that of Ru/SiO2 catalyst. The TOF of Ru–P/SiO2 catalyst increased with reduction temperature, and this TOF was lower than that of Ru/SiO2 catalyst. After HDN reaction, the peak of particle size distribution for Ru/SiO2 catalyst shifted to larger diameter, whereas that of Ru–P/SiO2 catalyst remained the same. Therefore, the stable activity of Ru–P/SiO2 catalyst can be explained by excess phosphorus species acting to stabilize Ru2P particles.View full abstractDownload PDF (1026K)
JOURNAL FREE ACCESSCatalytic properties of ruthenium phosphide (Ru2P) supported on silica for hydrodenitrogenation (HDN) of pyrrole were compared with those of Ru/SiO2 catalyst to clarify the effect of phosphidation. At higher W/F (652 g h mol−1), pyrrole conversion over Ru/SiO2 catalyst remarkably decreased with increasing reduction temperature. In contrast, pyrrole conversion over P-added Ru (Ru–P/SiO2) catalyst at the same W/F was enhanced with increasing reduction temperature due to Ru2P formation. This activity was higher than that of sulfided NiMoP/Al2O3 catalyst but lower than that of Ru/SiO2 catalyst at the same W/F. At lower W/F (130-391 g h mol−1), Ru–P/SiO2 catalyst showed higher activity and stability for pyrrole HDN than Ru/SiO2 catalyst. The cracking products (almost all CH4) were formed over Ru/SiO2 catalysts and butanes were formed over Ru–P/SiO2 catalysts. The results of CO adsorption and TEM images revealed that the particle size of Ru–P/SiO2 catalyst was smaller than that of Ru/SiO2 catalyst. The TOF of Ru–P/SiO2 catalyst increased with reduction temperature, and this TOF was lower than that of Ru/SiO2 catalyst. After HDN reaction, the peak of particle size distribution for Ru/SiO2 catalyst shifted to larger diameter, whereas that of Ru–P/SiO2 catalyst remained the same. Therefore, the stable activity of Ru–P/SiO2 catalyst can be explained by excess phosphorus species acting to stabilize Ru2P particles.View full abstractDownload PDF (1026K)
Letter
-
Masato Taki, Akira YoshidaArticle type: Letter
2013 Volume 56 Issue 2 Pages 102-103
Published: 2013
Released on J-STAGE: May 01, 2013
 JOURNAL FREE ACCESSFree sulfur is often detected when the flame photometric detector gas chromatography is used to analyze the bilge (oily mixture accumulated in the bottom of a ship). In the determination of the source of the discharge, the free sulfur peak interferes with the identification analysis. When a method to remove the free sulfur using the copper and sulfur reaction to form copper sulfide was considered, the free sulfur was removed satisfactorily. The result indicates that this method is effective for the removal of the free sulfur.View full abstractDownload PDF (663K)
JOURNAL FREE ACCESSFree sulfur is often detected when the flame photometric detector gas chromatography is used to analyze the bilge (oily mixture accumulated in the bottom of a ship). In the determination of the source of the discharge, the free sulfur peak interferes with the identification analysis. When a method to remove the free sulfur using the copper and sulfur reaction to form copper sulfide was considered, the free sulfur was removed satisfactorily. The result indicates that this method is effective for the removal of the free sulfur.View full abstractDownload PDF (663K) -
Hideo Nagata, Tomomi Ohta, Yasuyoshi Higuchi, Chisato Tanaka, Haruki M ...Article type: Letter
2013 Volume 56 Issue 2 Pages 104-107
Published: 2013
Released on J-STAGE: May 01, 2013
 JOURNAL FREE ACCESSThe hydrolysis of tetrafluoromethane (PFC-14) was studied over alumina-zirconia catalysts with varying alumina contents, prepared from boehmite. The initial PFC-14 conversion values were observed to increase with increasing catalyst alumina content. The alumina-zirconia catalyst used in this work possesses two types of acidity, termed (I) and (II), and previous work has shown that the hydrolysis of chloropentafluoroethane proceeds at a faster rate over acidity (I). The values of initial PFC-14 conversion in this work exhibited a positive correlation with the amount of acidity (I) in the catalyst. The hydrolysis of hexafluoroethane (PFC-116) was also investigated using these same catalysts and its initial conversion was found to peak at an alumina content of 90 wt% and to correlate with the total acidity (types (I) and (II) combined) of the catalyst. The catalytic cracking of isopropylbenzene is known to occur over the Brönsted acid sites of the catalyst, and the catalysis of isopropylbenzene in the presence of water vapor over two different catalysts indicated that acidity (I) corresponds to Lewis acidity and acidity (II) to Brönsted acidity. Accordingly, it is concluded that PFC-14 is decomposed by Lewis acid sites, whereas PFC-116 is decomposed by both Lewis and Brönsted acid sites.View full abstractDownload PDF (839K)
JOURNAL FREE ACCESSThe hydrolysis of tetrafluoromethane (PFC-14) was studied over alumina-zirconia catalysts with varying alumina contents, prepared from boehmite. The initial PFC-14 conversion values were observed to increase with increasing catalyst alumina content. The alumina-zirconia catalyst used in this work possesses two types of acidity, termed (I) and (II), and previous work has shown that the hydrolysis of chloropentafluoroethane proceeds at a faster rate over acidity (I). The values of initial PFC-14 conversion in this work exhibited a positive correlation with the amount of acidity (I) in the catalyst. The hydrolysis of hexafluoroethane (PFC-116) was also investigated using these same catalysts and its initial conversion was found to peak at an alumina content of 90 wt% and to correlate with the total acidity (types (I) and (II) combined) of the catalyst. The catalytic cracking of isopropylbenzene is known to occur over the Brönsted acid sites of the catalyst, and the catalysis of isopropylbenzene in the presence of water vapor over two different catalysts indicated that acidity (I) corresponds to Lewis acidity and acidity (II) to Brönsted acidity. Accordingly, it is concluded that PFC-14 is decomposed by Lewis acid sites, whereas PFC-116 is decomposed by both Lewis and Brönsted acid sites.View full abstractDownload PDF (839K)
- |<
- <
- 1
- >
- >|