Volume 62, Issue 1
Displaying 1-20 of 20 articles from this issue
- |<
- <
- 1
- >
- >|
Overview
-
Article type: Overview
2021 Volume 62 Issue 1 Pages 1-15
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 30, 2020 Download PDF (3137K) Full view HTML
Download PDF (3137K) Full view HTML
Regular Article
Materials Physics
-
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 16-19
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 30, 2020Download PDF (793K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 20-26
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: December 04, 2020 Download PDF (4004K) Full view HTML
Download PDF (4004K) Full view HTML
Microstructure of Materials
-
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 27-33
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
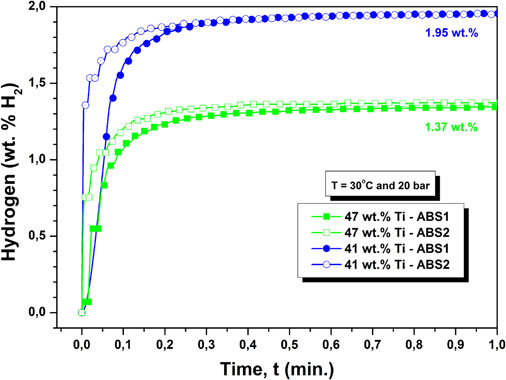 Download PDF (3720K) Full view HTML
Download PDF (3720K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 34-40
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 20, 2020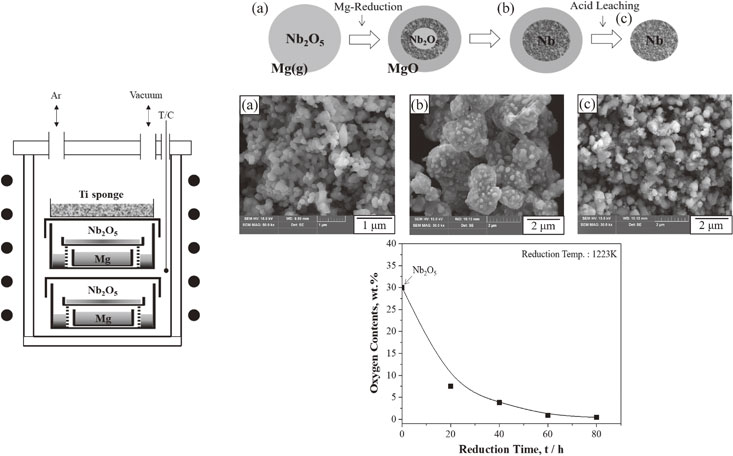 Download PDF (4266K) Full view HTML
Download PDF (4266K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 41-47
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 20, 2020 Download PDF (2575K) Full view HTML
Download PDF (2575K) Full view HTML
Mechanics of Materials
-
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 48-56
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 13, 2020Download PDF (4349K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 57-61
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 30, 2020 Download PDF (5359K) Full view HTML
Download PDF (5359K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 62-68
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: December 04, 2020Download PDF (5642K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 69-74
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 13, 2020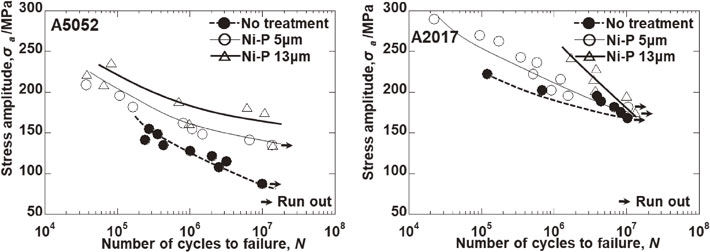 Download PDF (4241K) Full view HTML
Download PDF (4241K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 75-81
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 30, 2020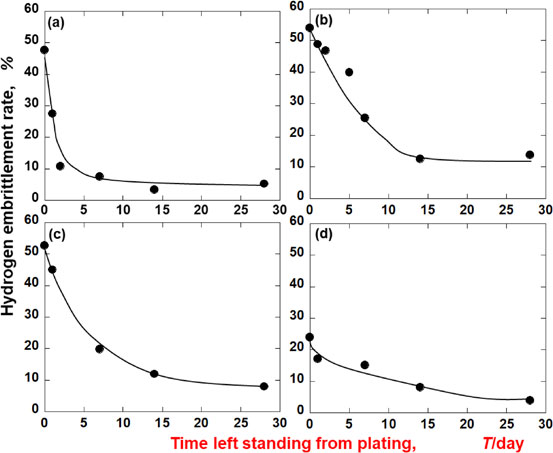 Download PDF (3376K) Full view HTML
Download PDF (3376K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 82-87
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
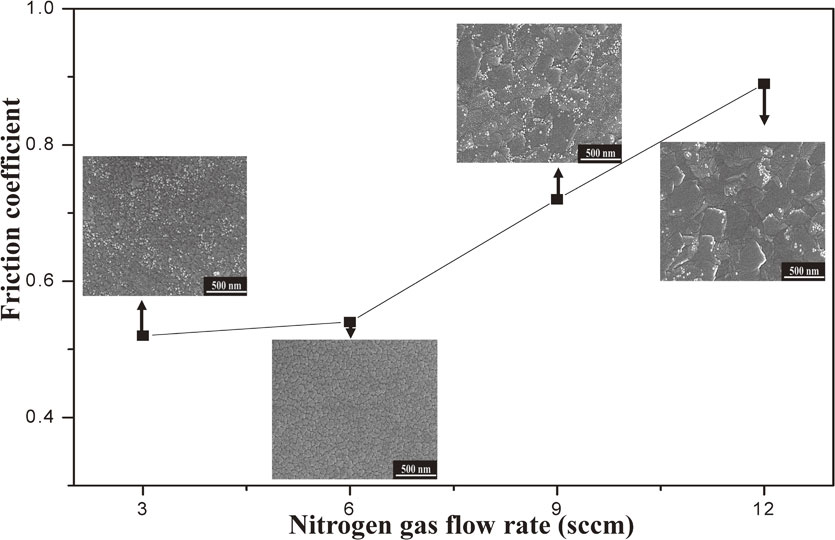 Download PDF (4154K) Full view HTML
Download PDF (4154K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 88-97
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 30, 2020 Download PDF (5539K) Full view HTML
Download PDF (5539K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 98-104
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
 Download PDF (5048K) Full view HTML
Download PDF (5048K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 105-110
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: December 04, 2020 Download PDF (1900K) Full view HTML
Download PDF (1900K) Full view HTML
Materials Chemistry
-
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 111-117
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 30, 2020 Download PDF (4663K) Full view HTML
Download PDF (4663K) Full view HTML
Engineering Materials and Their Applications
-
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 118-123
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 30, 2020 Download PDF (1735K) Full view HTML
Download PDF (1735K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 124-129
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 20, 2020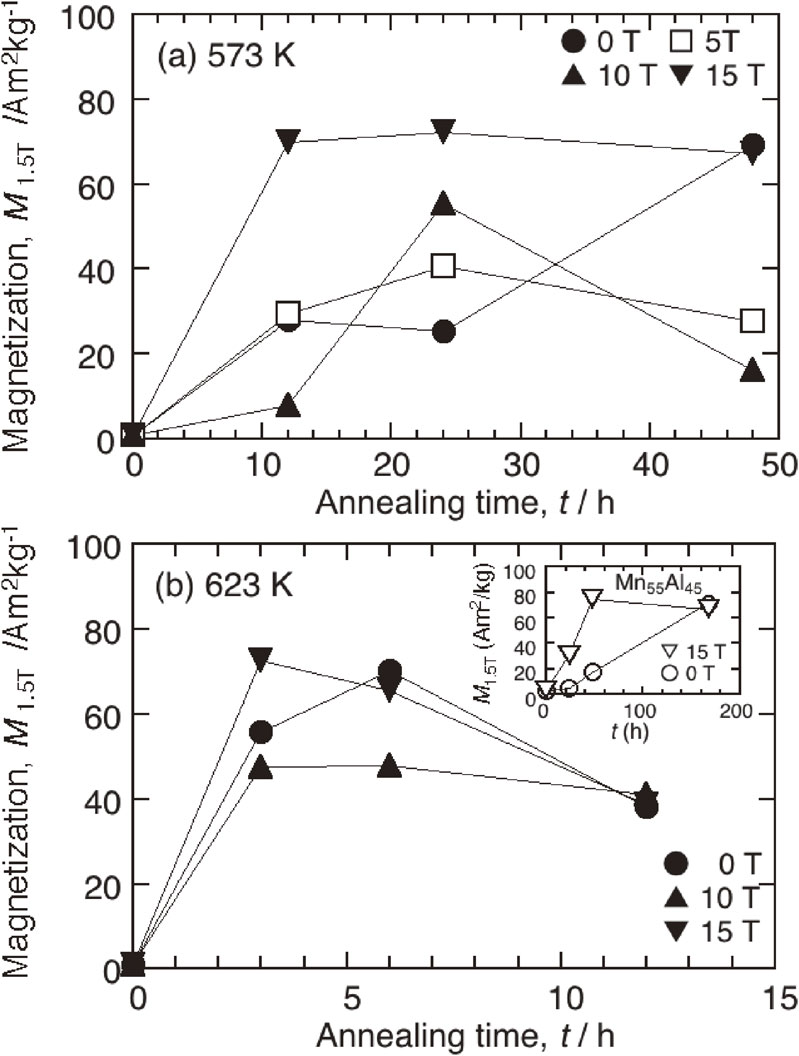 Download PDF (2793K) Full view HTML
Download PDF (2793K) Full view HTML -
Article type: Regular Article
2021 Volume 62 Issue 1 Pages 130-134
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
 Download PDF (2151K) Full view HTML
Download PDF (2151K) Full view HTML
Express Rapid Publication
-
Article type: Express Rapid Publication
2021 Volume 62 Issue 1 Pages 135-138
Published: January 01, 2021
Released on J-STAGE: December 25, 2020
Advance online publication: November 30, 2020 Download PDF (1905K) Full view HTML
Download PDF (1905K) Full view HTML
- |<
- <
- 1
- >
- >|