30th Symposium on Progress in Organic Reactions and Syntheses
Displaying 1-50 of 146 articles from this issue
October 19 (Tuesday), 2004
Oral Presentations
-
Session ID: 1O-01
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1O-02
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1O-03
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1O-04
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1O-05
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1O-06
Published: 2004
Released on J-STAGE: March 01, 2005

Poster Presentations
-
Session ID: 1P-01
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-02
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-03
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-04
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-05
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-06
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-07
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-08
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-09
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-10
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-11
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-12
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-13
Published: 2004
Released on J-STAGE: March 01, 2005
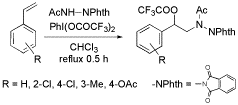
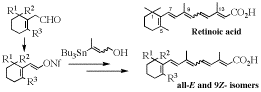
-
Session ID: 1P-14
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-15
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-16
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-17
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-18
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-19
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-20
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-21
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-22
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-23
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-24
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-25
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-26
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-27
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-28
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-29
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-30
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-31
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-32
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-33
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-34
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-35
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-36
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-37
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-38
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-39
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-40
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-41
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-42
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-43
Published: 2004
Released on J-STAGE: March 01, 2005

-
Session ID: 1P-44
Published: 2004
Released on J-STAGE: March 01, 2005