Fundamentals of High Temperature Processes
Thermodynamic Modeling of Liquid Steel
2020 Volume 60 Issue 12 Pages 2717-2730
Details
2020 Volume 60 Issue 12 Pages 2717-2730
Thermodynamic property of liquid steel is often described by activity coefficient of solute in the steel (the other form of partial excess Gibbs energy of the solute). Reliable description of the activity coefficient is required in order to predict equilibrium content of the solute as accurately as possible. In the present article, a number of such approaches are reviewed, with emphases on basic assumption and inherent character of each formalism/model, and on its applicability at high alloyed liquid steel. Chemical interaction between elements was categorized as weak interaction (i.e., between metal and metal) and strong interaction (i.e., between metal and non-metal). Each formalism/model was analyzed in the view of thermodynamic consistency (Gibbs-Duhem equation and Maxwell’s relation). It is concluded that two issues should be explicitly and simultaneously considered: obeying thermodynamic consistency and treating strong chemical interaction. The former ensures its applicability at higher solute content, and the latter is necessary to properly handle the strong interaction between metallic elements and non-metallic elements, contrary to conventional random mixing assumption.
This article provides a review of thermodynamic descriptions (theoretical model and formalism) of liquid steel which is basically an Fe based multicomponent liquid alloy. This is an extension of excellent thermodynamic analyses of liquid alloy by Darken,1,2) Schuhmann Jr.,3) and Pelton.4) Several thermodynamic models and formalisms have been used to calculate equilibrium solubility of various elements in steel in contact with other phases (gas, inclusion, slag, refractory, etc.). This article does not intend to provide a parameter set of particular model/formalism, but to show inherent character of the model/formalism as simple as possible. Moreover, beyond typical applications of those model/formalism confined in dilute region of liquid steel, it is also intended to discuss capability of those at higher content of elements in the steel. This would be useful when high alloyed steel such as TRIP, TWIP, and lightweight steel is developed.
Thermodynamic property of liquid steel has been often treated by formulating activity coefficient of component (γi) in liquid steel:
| (1) |
| (2) |
| (3) |
The activity coefficients (γi or fi) have been usually expressed as functions of composition and temperature. These are then used to calculate equilibrium content of each element under controlled activity in equilibrium with other phases. For example, a deoxidation equilibria in liquid steel by Al can be analyzed by:
| (4) |
| (5) |
At a given K(4) and
· thermodynamic consistency of models and formalisms
· applicability of the models and the formalisms to higher content beyond dilute region
· ability of the models and the formalisms in treating of strong interaction among components
Throughout this article, components of liquid steel are designated by 1, 2, ∙∙∙, N-1, and N (1 is a solvent) or Fe–M–X where M is a metallic component and X is a non-metallic component.
G (or Gex) stands for total energy (or the excess Gibbs energy) of the steel in J, while g (or gex) stands for the molar Gibbs energy (or the molar excess Gibbs energy) in J mol−1. γi can be obtained either from Gex or gex via:
| (6) |
Liquid steel is an Fe-based multicomponent liquid alloy. Typical elements of liquid steel are listed in Table 1. These elements may be classified by the following characters:
· metallic vs non-metallic
· stable as condensed phase (liquid or solid) vs stable as gas phase at steelmaking temperature
· attractive to Fe vs repulsive to Fe
| M | O(g) | S(g) | P(g) | Si(l) | B(l) | Al(l) | Ti(l) | Ta(s) | Nb(s) | V(s) | Y(l) | W(s) | Ni(l) | Cr(s) |
| ΔH (kJ mol−1) | −230 | −68 | −48 | −42 | −33 | −21 | −19 | −14 | −13 | −9 | −8 | −5 | −5 | −4 |
| −12.5 | −3.3 | 8.4 | 13.2 | 2.5 | 5.6 | 2.7 | −0.7 | 0.2 | 0 | |||||
| M | Mo(s) | Mn(l) | Ce(l) | La(l) | N(g) | Sn(l) | Cu(l) | Zn(g) | C(s) | H(g) | Mg(g) | Ca(g) | Pb(g) | |
| ΔH (kJ mol−1) | −3 | −1 | −1 | 0 | 7 | 8 | 16 | + | + | + | + | |||
| 0 | 0.8 | 7.1 | 6 | 6.6 | 1 |
H, B, C, N, O, P, S are regarded as the non-metallic elements among the components listed in the Table 1. H, N, O, P, S as well as Ca, Mg, Pb, Zn vaporize at temperature of steelmaking. Therefore, solubility of these elements in liquid steel are generally low. Interaction with Fe (either attractive or repulsive) was characterized by the enthalpy of mixing (ΔH). Among those elements, O shows the most negative ΔH, representing very strong attraction to Fe. On the other hand, Ca and Mg are known to form wide immiscibility with Fe, although accurate ΔH could not be measured due to its volatile character. In general, non-metallic element exhibits strong attraction to Fe (and other metals), although some exceptions are found (H, N). The strong attraction between elements requires careful description of the activity coefficient (or excess Gibbs energy), due to significant deviation from random mixing behavior of atoms.
Chipman and coworker noticed that activity coefficient of a solute in liquid steel depends on significantly by C content.5,6,7,8,9) For example, 2 mass pct. of C increases the activity coefficient of Si by 2 times. This necessitates a formulation of the activity coefficient as a function of composition (apart from temperature). Wagner employed a Taylor series expansion for the ln γi.10) This yields a well-known Wagner’s Interaction Parameter Formalism (WIPF):
| (7) |
Using the Maxwell relations, Wagner showed that:
| (8) |
| (9) |
Extensive literature survey resulted in a compilation of the interaction parameters.12,13,14)
It is stressed that the evaluation of each parameter was performed independently giving the best fit to the experimental data within limited composition and temperature range. Since this is a mathematical formalism based on the fact that the γi should be a function of composition5,6,8,9) at a given temperature, it does not ensure that the γi of the WIPF obeys thermodynamic consistency condition, as will be discussed in Sec. 4.1.1,3,4,;15,16,17, 18,19,20,21,22,23,24,25) Although its definition is strictly valid for the limiting case (Xi → 0 for all i),10) it has been often used at finite content with some success. Although it is easy to use and widely applied in metallurgical community, its validity is somewhat limited. It often fails in calculation of equilibrium content of solutes in liquid steel, when it contains strong deoxidizer or is applied to higher solute content. In the following sections, further efforts to overcome this shortcoming of WIPF are reviewed. In order to deliver the content of the present article efficiently, characteristics of the activity coefficients of various solutes in liquid steel are introduced in advance.
In 1–2 binary system showing negative deviation, the γ2 is less than unity. It increases as X2 increases, and approaches to the unity at X2 = 1 (Raoult’s law). Most simple and natural increase of the ln γ2 may be represented schematically in Fig. 1(a) (Case I). The ln γ2 increases monotonously as X2 increases. This may be represented by a simple regular solution model:
| (10) |
| (11) |
| (12) |

Activity coefficient, excess Gibbs energy, and excess stability of 1–2 binary solution: (a)–(c) weak interaction between 1 and 2, (d)–(f) strong interaction between 1 and 2, (g)–(i) strong interaction between 1 and 2, with a repulsion between 1 and “12”.
When the interaction becomes stronger (Case II), the ln γ2 increases as X2 increases, but somewhat in different manner, as seen in Fig. 1(d): the ln γ2 increases moderately at low X2, then increases suddenly at a particular composition (X2 = ~0.5 in this example), followed by approaching moderately to zero (by the Raoult’s law). This is interpreted as follows. Adding small amount of 2 in pure 1 results in strong ordering of the 2. This lowers the activity of 2 compared to that in a weak interacting system (or random mixing solution). Therefore, the ln γ2 increases only slightly while X2 increases. This continued up to the composition of maximum ordering where the solvent 1 could bind 2 as much as possible. Over this composition, additionally added 2 becomes free from the binding by 1, thereby a2 increases as if 2 were in an ideal solution (ln γ2 = ~0). The strong ordering at low X2 results that gex (mostly enthalpy of mixing) changes as much as 2 enters in the solution. Contrary to the parabolic shape (Fig. 1(b)), a sharp decrease of the gex is expected (linearly proportional to X2), yielding a “V-shape” (Fig. 1(e)). This is consistent with the “titration-like” curve of the ln γ2 (Fig. 1(d)). Corresponding Ψ shows a pronounced peak near X2=0.5, implying a strong ordering at the composition (Fig. 1(f)).
Both two cases (Case I and Case II) are representing negative deviation, therefore ln
Change of the activity coefficient may be qualitatively read from the phase diagram. Figure 2 shows phase diagrams of Fe–P, Fe–S, and Fe–O binary systems (Fe: 1, P/S/O: 2 in the above analysis). P, S, and O exhibit very strong attraction to Fe (ΔH < 0), thereby yielding negative deviation (γ2 < 1), as listed in the Table 1.
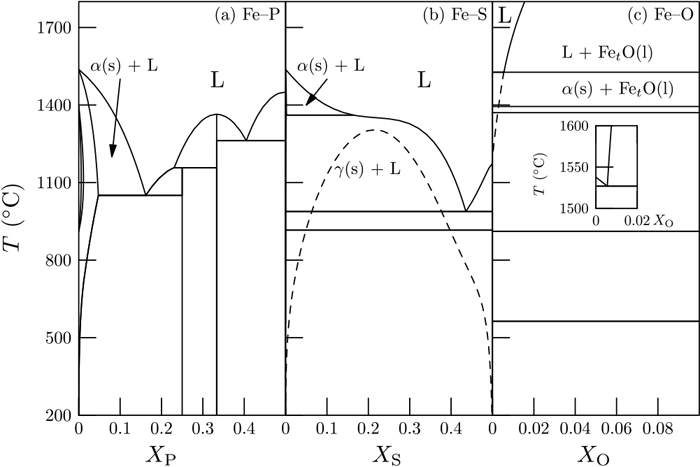
Binary phase diagram of (a) Fe–P, (b) Fe–S, and (c) Fe–O (with an inset of a part of the phase diagram near the melting temperature of Fe) systems.
The activity coefficient of components in liquid steel is basically a result of thermodynamic characteristic of chemical interaction between the components (not only with Fe but also with other solutes). Different signs of
Darken identified some regularities of ln γ1 and ln γ2 in a terminal region in 1(solvent)–2(solute) binary liquid solution. At a given T, he found that the ln γ1 is adequately represented by the following equation:
| (13) |
When the Gibbs-Duhem equation
| (14) |
| (15) |
| (16) |

The above expression is inherently a second order interaction parameter formalism. However, contrary to the WIPF, the first- and second-order parameters are interdependent. When the Eq. (16) is compared to the Eq. (9) in the 1–2 binary system, (α12 + I12) corresponds to ln
| (17) |
Darkens extended his quadratic formalism to a ternary 1–2–3 system:2)
| (18) |
The last term was specially designed in order to yield a ternary regular solution model:
| (19) |
| (20) |
| (21) |
| (22) |
When the Eqs. (20), (21), (22) are compared to the Eq. (9) in the 1–2–3 ternary system, it is easily seen that:
| (23) |
| (24) |
| (25) |
From the above relations, it can be seen that the interaction parameters (εij, ρij, ρij,k) are indeed dependent each other. Lupis and Elliott provided more general relationships among the interaction parameters.11) From the above analysis, it is evident that
· I12 and I13 must be zero if the above formalism is to be valid even at X2 → 1 (and X3 → 1).
· the Eqs. (23), (24), (25) should be satisfied in order for this formalism to be thermodynamically consistent.
Darken claimed that WIPF is not thermodynamically consistent except at infinite dilution.1) This has been well appreciated by numerous publications.3,4,15,16,17,18,19,20,21,22,23,24,25) For the activity coefficient as the partial excess Gibbs energy, the Gibbs-Duhem equation must be satisfied:
| (26) |
Also, the following equation must satisfy in order to fulfill the thermodynamic consistency in 1–2–3 ternary solution:2)
| (27) |
This is basically obtained by the Maxwell relation:
| (28) |
The two Eqs. (26) and (27) must be satisfied for the activity coefficients in the 1–2–3 ternary solution in order to keep thermodynamic consistency condition.2)
4.2. Unified Interaction Parameter FormalismUsing the Eqs. (23), (24), (25), the Eqs. (20), (21), (22) can be rewritten as1:
| (29) |
| (30) |
| (31) |
The terms in the parentheses are identical to the ln γ1, regardless of the solutes. It is to be noted that the activity coefficient of solvent was not considered in WIPF. Pelton and Bale recognized that if one wants to use the Eq. (9) but in a thermodynamically correct way, the second-order terms in the Eq. (9) should be formulated as seen in the Eqs. (30) and (31).35,36) This is indeed the same as adding the ln γ1 to the ln γ2 (and the ln γ3) of the first-order term (first two terms in the Eq. (9)):
| (32) |
| (33) |
They showed that the Eqs. (32) and (33) satisfies the Gibbs-Duhem equation (Eq. (26) with the ln γ1 (Eq. (29)) and the Maxwell relation (27). This ensures necessary and sufficient thermodynamic consistency, and eliminates the errors in the WIPF. Moreover, well evaluated first-order interaction parameters of the WIPF can be directly used without any conversion.12,13,14) They call this formalism as Unified Interaction Parameter Formalism (UIPF), because this unifies the WIPF at the infinite dilution, Lupis and Elliott’s second order formalism, Darken’s formalism at higher solute content.
The UIPF can be expanded to include second-order parameters (εijk in the notation of Pelton and Bale4,35,36)) or higher-order parameters for the activity coefficient of solutes in 1–2–∙∙∙–N multicomponent solution. This satisfies the Gibbs-Duhem equation in the N–component system (Eq. (14)) and the Maxwell relation such as the Eq. (28) for all solutes i and j (i, j = 2, ∙∙∙, N).
Pelton and Bale proposed that the addition of ln γ1 to the conventional WIPF is the way of correction of the Wagner’s formalism to be thermodynamically consistent.4,35,36) This ensures that the application of interaction parameter formalism even at higher content of solutes is thermodynamically correct.
Malakhov raised a possibility that there may be infinite number of ways of such correction to the WIPF, which can satisfy both the Gibbs-Duhem equation (Eq. (26)) and the Maxwell relation (Eq. (27)) in 1–2–3 ternary solution.24) However, recently, the present author pointed out that considering the Maxwell relation not only between solute-solute but also between solvent-solute resolves the issue raised by Malakhov, and concluded that the correction made by Pelton and Bale is the only way of such corrections.25) The present author extended the Eq. (27) to the N-component system including the solvent-solute interaction:
| (34) |
Miki and Hino applied the Darken’s quadratic formalism to interpret deoxidation phenomena in high alloyed steel.37,38,39,40,41,42,43,44) They expanded the interaction energy (ωij) using Redlich-Kister type polynomial.45) This yields essentially identical formalism to the UIPF up to higher order, or the proposal by Hillert using a modified regular solution model for terminal solution.46)
4.3. Regular Solution for Dilute Component XConsider that a non-metallic component X dissolves in a binary Fe–M metallic liquid solution (Fe–M–X). As mentioned in Sec. 1, interaction between Fe–M is generally weaker than that between Fe–X and M–X. Alcock and Richardson treated the dissolution of the X in the Fe–M binary solution in a pairwise manner, and counted number of pairs and associated energy change upon the dissolution.47) They derived the following equation for the Henrian activity coefficient of infinitely dilute solution of X (
| (35) |
| (36) |
| (37) |
| (38) |
| (39) |
The Eq. (39) is indeed equivalent to the Eq. (25). The Eq. (35) can be easily extended to multi-component 1–2–∙∙∙– (N−1) –X.47)
4.4. DiscussionIt can be concluded that Darken’s formalism in Sec. 4.1,1,2) Unified Interaction Parameter Formalism in Sec. 4.2,4,35,36) Hillert’s modified regular solution model,46) and Alcock and Richardson’s regular solution model in Sec. 4.347) are essentially identical inasmuch as these are all based on the well known regular solution theory. Darken’s starting point (Eq. (13)) indeed conforms with the regular solution theory, of which the energy change is basically described by pairwise interaction. It should be stressed that in this theory, a probability of finding a nearest pair of (i–j) is 2XiXj when i ≠ j or Xi2 when i = j. This is equivalent to say that the atoms i and j are distributed randomly over a quasi-lattice, regardless of the size of interaction energy ωij (or αij). It is evident that the interaction between metal and non-metal cannot be successfully described assuming such näive random mixing concept. Alcock and Richardson found that their model (Eq. (35)) works well when a difference between ln


Test calculation of the regular solution model: (a) ln γM and (b) ln γ°X in liquid Fe–M–X alloy with various αFeM, at infinite dilute X.
Alcock and Richardson48) and Jacob and Alcock59) subsequently proposed a quasichemical approach in which preferential formation of nearest neighbor pair was explicitly considered in order to consider such a non-random behavior of atoms. They showed somewhat improved results compared to the original regular solution model (Eq. (35)) which assumes random distribution of atoms.
Fe–S binary liquid may be chosen as one of examples exhibiting strong interaction. Figure 6 shows the enthalpy of mixing in liquid Fe–S alloy as a function of XS.60,61) This is a half of “V-shape” curve in Fe-rich side, demonstrating very strong attraction between Fe and S. A slight positive deviation from the rigorous “V-shape” (straight line) is due to the repulsion between Fe (with very low S) and S (coupled with Fe), yielding
Chipman interpreted this phenomena as if the S entering into the liquid Fe forms a strong and stable bond,62) similarly done by Belton and Tankins for Fe–O and Fe–S alloys.63) He showed convincingly the need for alternative definition of new composition variable. The following is an interpretation of this phenomenon by Schuhmann3) that the activity coefficient of strong interacting solute X (mostly non-metallic component such as S in liquid Fe) can be well represented by a model which considers “M” and “MX” as components, where the “MX” is formed by the following reaction:
| (40) |
| (41) |
| (42) |
Activities of the species are then defined as:
| (43) |
| (44) |
| (45) |
| (46) |
From the equilibrium constant K(40), the activity of S is obtained by inserting the Eqs. (41) and (42) into the Eqs. (43) and (44):
| (47) |
By setting γ′X = (1/K(40))(γ′MX/γ′M) and substituting the Eqs. (45) and (46), the ln γ′X can be expressed as:
| (48) |
Chipman proposed to use the following composition variables:62,64,65)
| (49) |
It is seen that X’MX = yX in this particular case. Therefore, the activity of X in the Fe–M–X alloy is defined with the new “lattice ratio” zX (aX = γ′XzX) and the activity coefficient of X is expressed as a first-order interaction parameter formalism of “atom ratio”:
| (50) |
This formalism is thermodynamically consistent and useful in describing the activity coefficient of nonmetallic solutes in liquid steel. Ban-ya and co-worker successfully applied this formalism in the description of gas element solubility in liquid steel and alloy.9,66,67,68,69)
However, this requires the use of unusual composition variables (zi and yi), which were not required in the previous formalism and models discussed in the Sec. 4.1 for weak interacting system. Nevertheless, this approach was successful in extending the regularity found by Darken in the weak interacting system (Eqs. (13) and (15)) to systems with strong interacting components such as non-metallic elements. It can be easily extended to any number of X.4) More importantly, this approach gives some insight that the choice of solution components imply a difference in structure of the solution, contrary to a random mixing solution. Consequently, the entropy of mixing in the liquid steel containing both metallic element and non-metallic element shall be different to the ideal entropy of mixing.
5.2. Associate ModelWithout introducing the new composition variables, the excess Gibbs energy may be described by explicitly considering possible “associate”. The previous example may be regarded as a system composed of M, X, and MX where the first two represent unassociated species and the last means an associate. Mole fraction of the species (X’i) can be used which must be balanced with atom fraction (Xj). This approach has been applied several cases, showing significant improvement in treating both weak interaction and strong interaction in liquid steel. This idea was used by Chipman and co-workers,70,71) Belton and Tankins,63) and Zapffe and Sims.72) Wasai and Mukai applied this concept in the Fe–Al–O system by considering all possible associates: Fe3Al, FeAl, FeAl3, FeO, FeAl2O4, Al2O3, Al2O.73,74) These associates were selected from the compounds found in the phase diagram of Fe–Al, Fe–O, and Fe–Al–O system. Assuming the ideal mixing among the species – the associates and unassociated species – equilibrium composition of all the species were calculated when the liquid is in equilibrium with a solid Al2O3. Bouchard and Bale treated this solution a bit simpler by only considering O-containing associates (AlO and Al2O).75) They considered total five species (Fe, Al, O, AlO, and Al2O) in the framework of UIPF. The following mass balance equations must be kept:
| (51) |
| (52) |
| (53) |
The following associate formation reactions are considered:
| (54) |
| (55) |
| (56) |
| (57) |
Since the strong attraction between Al and O was treated by the presence of two associates, interaction between each species can be considered as the weak interaction or close to ideal mixing. Therefore, the γi was formulated by the UIPF for weak interaction. The internal equilibrium must be solved by using the Eqs. (51), (52), (53), (56), and (57) in order to obtain the equilibrium amount of all the five species. This gives the activities of real elements Al and O (aAl = γ′AlX′Al and aO = γ′OX′O).
For deoxidation equilibria in liquid steel containing Al and O, the aAl and aO must satisfy the following equilibrium constant equation for the heterogeneous equilibrium between the liquid steel and the solid Al2O3:
| (4) |
| (5) |
Standard state of Al and O may be chosen to be the Raoultian or the Henrian standard state for convenience, which must be reflected in K(4). After solving the heterogeneous equilibrium, the equilibrium amount of all the species in the liquid steel are then back substituted in the Eqs. (51), (52), and (53) in order to obtain final equilibrium composition of the steel. Jung et al. extended this model to other steel system showing strong ordering between M and O, assuming the Henrian behavior of all O containing associates:76)
| (58) |
It was shown that this approach could explain deoxidation equilibria in the liquid steel containing various alloying elements in Fe-rich region, better than the usual approach using WIPF that inherently assumes random mixing between atoms. However, this approach was limited to Fe-rich region due to the Henrian activity coefficients of various alloying elements (ln
Wagner proposed that a nonmetallic element is likely to dissolve in liquid metal interstitially.77) He developed a model which describes ln
| (59) |
| (60) |
| (61) |
The probability of finding (Z – i) Fe atoms and i M atoms randomly surrounding a vacant site, regardless of configuration of the Fe and the M, is:
| (62) |
| (63) |
Therefore, total atom fraction of X (XX) can be obtained by summation of the
The Reaction (59) reads as a general chemical reaction:
| (64) |
By taking pure X2(g) at 1 bar as the standard state of X, and assuming the Henrian behavior of X, the following is obtained:
| (65) |
Therefore, summation of the Eq. (63) from i = 0 to Z, and substituting it into the Eq. (65) along with the Eq. (61) yields:
| (66) |
| (67) |
When it is compared with the Eq. (39), Z and h appear. Wagner proposed to use Z = 6, in analogy of octahedral interstitial sites of a close-packed hexagonal lattice or that of a fcc lattice.77) h, from the definition in Eq. (61), is somewhat linked to αFeM in the Eq. (39), the interaction between Fe and M. An empirical equation for the h has been proposed.78) This model have been applied to describe dissolution of O, S, N78,79,81,83,89,90) in various alloys and extended to multicomponent system.80)
5.4. DiscussionThe two models shown in this section were used to calculate
| Model | Structure | Parameters | Note |
|---|---|---|---|
| UIPF (regular solution model) | Fe, M, X | ln γX(Fe) = 0, ln γX(M) = −15, αFeM = −5, −2, 0, 5, 10, 30 | Fig. 5 (a), (b) |
| UIPF with associate | Fe, M. X, MX | ln | Fig. 7 (a), (b) |
| Solvation shell model | V(FeZ-iMi), M(FeZ-iMi) | Z = 6, ln γX(Fe) = 0, ln γX(M) = −15, h = 7.26 kJ | Fig. 7 (c), (d) |
| Modified Quasichemical Model | Fe, M, X | Z = 6, ΔgFeM = −10 kJ, ΔgFeX = −20 kJ, ΔgMX = −100 kJ, | Fig. 7 (e), (f) |

Test calculations for ln γM and ln
The γM does not show a large positive deviation from the ideality, contrary to the previous example calculation in Fig. 5. The model calculations in Fig. 7 look more reasonable that the stronger ordering between M and X is taken into account regardless of the interaction between Fe and M.
In case of the UIPF with the associate,
The solvation shell model assumes the Henrian behavior of X in the liquid Fe–M alloy, therefore the
Various approaches were discussed in Sec. 4 and Sec. 5. It was shown that two important issues should be explicitly taken into account: thermodynamic consistency and strong SRO between elements, in particular between metal and non-metal. The formalism and the models in Sec. 4 satisfy the thermodynamic consistency (Eqs. (14), (34)), but are inherently assuming random mixing among components. The formalism and the in Sec. 5 were designed to the treat strong SRO in liquid steel. However, introducing new composition variable (Sec. 5.1), or ln
In this section, an improved approach is shown which satisfies both the thermodynamic consistency and the SRO, up to higher concentration of metallic component. In order to describe the Gibbs energy of liquid steel, which takes into account both thermodynamic consistency at high metal content and strong SRO, two thermodynamic models can be mentioned. One is the Central Atoms model developed by Lupis and Elliott,93,94) and is essentially identical to “Surrounded Atom model” developed by Mathieu and co-workers independently.95) Basic species of this model is a cluster of an atom and its nearest neighbor shell, similar to the Wagner’s solvation shell model.77) The central atom may be a substitutional atom or an interstitial atom in case of the steel. This model is not limited to Henrian region of nonmetallic element. Lehmann and co-worker extended this approach to treat both the steel and the slag and called it Generalized Central Atom Model.96,97) This model has been successfully used in a computing software CEQCSI.
The other is the Modified Quasichemical Model (MQM), which was developed by Pelton and co-worker.98,99,100) Basic species of this model is either a nearest-neighbor pair98) or a quadruplet composed of two sites for metal and two sites for non-metal or vacancy.100) The pair approximation of this model is relatively simpler than the quadruplet approximation of this model100) and the Central Atoms Model,93,94) but it explicitly takes into account the SRO and keeps thermodynamic consistency.
In a Fe–M–X ternary solution, six different nearest-neighbor pairs are considered by the following reactions:
| (68) |
| (69) |
| (70) |
| (71) |
| (72) |
| (73) |
The Gibbs energy of this solution is then described by:98)
| (74) |
| (75) |
The equilibrium number of moles of the six i-j pairs (nFeFe, nFeM, nFeX, nMM, nMX, and nXX) in the solution of a composition (fixed nFe, nM, and nX) at a given T and P are obtained at an internal equilibrium by setting:
| (76) |
From the Eq. (76), the following quasichemical equilibrium constants are obtained:
| (77) |
| (78) |
| (79) |
From the six equations (3 mass balance equations (Eqs. (71), (72), (73))) and 3 quasichemical equilibrium constant equations (Eqs. (77), (78), (79))), the six unknowns (nFeFe, nMM, nXX, nFeM, nFeX, and nMX) can be obtained. (In N-component system, there are N(N + 1)/2 equations and unknowns, respectively). Substituting the nij back to the Eqs. (74) and (75) gives the Gibbs energy of the solution. This model can describe the Gibbs energy of a solution whose sub-system behaves as an ideal solution or weak-interacting solution or strong-interacting solution simultaneously. Activity coefficient of component can also be numerically obtained using the Eq. (6), although a heterogeneous equilibrium can be directly calculated by minimizing Gibbs energy of the whole system including the liquid steel and other phase (slag, inclusion, refractory, etc.). This model extends from pure metal Fe to pure metal M, and extends toward higher X content keeping the thermodynamic consistency. Therefore, this model is applicable to high alloyed steel with strong-interacting element X. This model easily extends to the multicomponent system,108) and has been successfully used with a computing software FactSage.109,110)
6.1. Discussionln
The MQM has been applied to Fe–Al–O, Fe–Mn–O, and Fe–Mn–Al–O systems by Paek et al.58,111) Deoxidation equilibria of the Fe–Al–O system are shown in Fig. 8: equilibrium content of Al and O in liquid steel in equilibrium with solid Al2O3. Excellent agreement with the available experimental data is seen at wider composition range up to pure liquid Al and wider temperature range. Such an agreement could not be obtained by other approaches reported before. This is an important improvement in describing thermodynamics of liquid steel, in particular for high alloyed steel such as TRIP, TWIP, and light weight steel. This model has been also applied to systems containing C and S.57,112,113,114,115,116)

Comparison between the deoxidation experimental data and the model calculation by MQM in the Fe–Al–O system. Reprinted from Paek et al.111) (Online version in color.)
Several formalisms and models for the representation of excess partial Gibbs energy (the logarithm of activity coefficient) or the integral Gibbs energy were reviewed. Widely used Wagner’s Interaction Parameter Formalism (Sec. 2) is simple to use, and many number of experimental researches were devoted to provide the interaction parameters. While it has enjoyed high popularity, its application was often limited due to inherent assumption of random mixing between atoms. Also the formalism is thermodynamically inconsistent except at the infinite dilution. In Sec. 3.1, interactions among components in liquid steel were analyzed, and the change of the activity coefficient by composition was discussed along with the phase diagram. The interactions were categorized as weak interaction and strong interaction. And the available formalisms and models were reviewed in the view of thermodynamic consistency and capability in treating the strong interaction.
In Sec. 4, Darken’s quadratic formalism, Unified Interaction Parameter Formalism, and regular solution model were reviewed. All of these approaches satisfy the necessary thermodynamic consistency (Gibbs-Duhem Eq. (14) and Maxwell relation (34)). However, when there is strong SRO between species, it does not give reasonable representation for the activity coefficient of the components. This is because these approaches inherently assume random mixing between atoms.
In Sec. 5, Chipman’s approach of using new composition variables (atom ratio yi and lattice ratio zi) was introduced. This is to explain strong attraction between metal atoms and non-metal atoms. While it showed better representation of the activity coefficient of the components, a general application to the multicomponent system is limited, where various metal atoms and non-metal atoms mix. By identifying that the use of the new composition variable was indeed identical to assume associates in the liquid, introducing several associates in the previous interaction parameter formalism has been proposed.73,74,75,76) It has been shown that this approach could be applied in a number of liquid steel systems, but it was only valid at dilute region of liquid steel.76) A solvation shell model developed by Wagner was a promising approach which describes the activity coefficient of non-metal atom X (ln
Sec. 6 referred two model approaches - Central Atoms Model and Modified Quasichemical Model. Both models were formulated for the integral Gibbs energy, which can be used to find the equilibrium by minimizing Gibbs energy of the whole system. Those can be applied at higher metal content, and consider the strong SRO by quasichemical approximation. In particular, the model description of the MQM in the pair approximation was introduced, and its application to steel deoxidation by Al, Mn, and Al–Mn was shown.58,111) It can be seen that the explicit consideration of thermodynamic consistency and strong SRO results in better agreement for the deoxidation equilibria. As the development of steel includes high alloyed steel such as TWIP, TRIP and lightweight steel, the thermodynamic modeling of liquid steel towards higher alloying content is now necessary to understand the chemical interaction in the steel and with other phases involved in steelmaking process such as slag, inclusion, refractory, etc.
