Abstract
Aim: Activin receptor-like kinase 7 (ALK7) acts as a key receptor for TGF-β family members, which play important roles in regulating cardiovascular activity. However, ALK7’s potential role, and underlying mechanism, in the macrophage activation involved in atherogenesis remain unexplored.
Methods: ALK7 expression in macrophages was tested by RT-PCR, western blot, and immunofluorescence co-staining. The loss-of-function strategy using AdshALK7 was performed for functional study. Oil Red O staining was used to observe the foam cell formation, while inflammatory mediators and genes related to cholesterol efflux and influx were determined by RT-PCR and western blot. A PPARγ inhibitor (G3335) was used to reveal whether PPARγ was required for ALK7 to affect macrophage activation.
Results: The results exhibited upregulated ALK7 expression in oxidized low-density lipoprotein (Ox-LDL) induced bone marrow derived macrophages (BMDMs) and mouse peritoneal macrophages (MPMs), isolated from ApoE-deficient mice, while ALK7’s strong immunoreactivity in BMDMs was observed. ALK7 knockdown significantly attenuated pro-inflammatory, but promoted anti-inflammatory, macrophage markers expression. Additionally, ALK7 silencing decreased foam cell formation, accompanied by the up-regulation of ABCA1 and ABCG1 involved in cholesterol efflux but the down-regulation of CD36 and SR-A implicated in cholesterol influx. Mechanistically, ALK7 knockdown upregulated PPARγ expression, which was required for the ameliorated effect of ALK7 silencing macrophage activation.
Conclusions: Our study demonstrated that ALK7 was a positive regulator for macrophage activation, partially through down-regulation of PPARγ expression, which suggested that neutralizing ALK7 might be promising therapeutic strategy for treating atherosclerosis.
Introduction
Cardiovascular diseases, associated with atherosclerosis, are recognized universally as the leading cause of mortality and morbidity worldwide
1)
. Atherosclerosis is widely accepted as a complex, multi-factorial disease, and chronic inflammatory disease, which can ultimately lead to major adverse cardiovascular events, including arteriosclerotic occlusive diseases, arteriovenous thrombosis, aneurysms, myocardial infarction, stroke, and sudden cardiac death
2)
. The endothelial dysfunction initiates the atherosclerosis process, characterized by secretion of chemokines and adhesion molecules to fasten the circulating monocytes to the endothelium
3)
. Monocytes, and the subsequently differentiated macrophages, play determinate roles in multiple aspects of atherogenesis
4)
. Macrophages are responsible for the inflammatory response in atherogenesis by regulating the balance between activation of classically pro-inflammatory M1 macrophages (analogous to the T-cell nomenclature T-helper cell 1 [Th1]), which is universally induced by a combination of interferon-γ (IFN-γ) with lipopolysaccharide, and, alternatively, anti-inflammatory M2 macrophages (analogous to Th2 T cells), which can be induced by IL-4 or IL-13
5-
8)
. Notably, the macrophages’ uptake of modified low-density lipoprotein (LDL) and subsequent transformation into foam cells continually contribute to development of atherosclerotic lesions and vulnerable plaques
9)
. Therefore, it is urgent to explore the key regulator of macrophage inflammation and foam cell transformation implicated in atherosclerosis.
The activation of transforming growth factor type-β (TGF-β) axis, from ligands, receptors, and binding proteins to ligand traps, plays an important role in regulating cardiovascular diseases
10,
11)
. As a key type I receptor for TGF-β family members, activin receptor-like kinase 7 (ALK7) has also emerged as the determinate receptor for nodal and activin, both of which are implicated in various diseases
12)
. In past decades, ALK7 signaling acted as a novel suppressor of tumorigenesis and metastasis, characterized by the formation of a homeostatic tissue barrier, which is identified as an effective approach for attenuating the attack from cancer cells and inhibiting the tumor’s capability for metastatic seeding
13,
14)
. Besides the effect on tumor development, ALK7 is specifically expressed during the late phase of adipocyte differentiation
15)
. The adipocytes’ specific ALK7 dysfunction causes increased lipolysis, which leads to decreased fat accumulation
16)
, while ALK7 is also associated with multiple factors implicated in metabolic disease
17)
. ALK7 decreases inflammatory adipocytokines secretion and improves glucose tolerance and insulin sensitivity
18)
. It also protects against the development of pathological cardiac hypertrophy
19)
. Notably, ALK7 activation initiates multiple downstream signaling pathways, including Smad family members, PI3K-AKT, Ras-Erk, and mitogen-activated protein kinases (MAPK)
19-
21)
, which are involved in atherogenesis. However, ALK7’s specific effect on the development of macrophage activation implicated in atherogenesis is unexplored.
In the present study, upregulated ALK7 expression was induced in macrophages upon oxidized low-density lipoprotein (Ox-LDL) treatment. The loss-of-function study demonstrated that ALK7 knockdown significantly alleviated pro-inflammatory, but promoted anti-inflammatory, macrophage polarization, accompanied by decreased foam cell formation. It enhanced ABCA1 and ABCG1 related to cholesterol efflux but reduced CD36 and SR-A involved in cholesterol influx. Mechanistically, we verified that PPARγ was required for the attenuated effect of macrophage activation, regulated by ALK7 knockdown.
Materials and Methods
Cell Culture and siRNA Transfection
Bone marrow derived macrophages (BMDMs) were isolated and harvested from the femurs and tibias of ApoE-deficient mice under sterile conditions. The nucleated bone marrow cells were collected from each mouse and then cultured in 10 ml of RPMI with 10% fetal bovine serum and MCSF (50 ng/ml). Mouse peritoneal macrophages (MPMs) were collected from ApoE deficiency mice via peritoneal lavage four days after the intraperitoneal injection of 4% thioglycolate (1 ml). Cells were cultured in RPMI containing 10% fetal bovine serum and 1% penicillin–streptomycin. The collected macrophages were seeded at 2x106/35-mm well in six-well plates. To knockdown ALK7 expression, ALK7 specific short hairpin RNA (shRNA)-expressing (shALK7) constructs, using a pENTR/U6-shRNA vector, were used to generate AdshALK7 recombinant adenoviral vectors, and short hairpin RNA (AdshRNA) served as controls. The BMDMs were transfected with the above adenovirus according to the manufacturer’s protocol. The cells were then stimulated with 15 ng/ml Ox-LDL, which was purchased from sigma (O0625–25g), for 24 hours after serum starvation for one day.
Quantitative Real-Time PCR
Total RNA from macrophages was isolated with TRIzol reagent and was held constant at 20 µl per tube in each experiment. Then the cDNA obtained via reverse-transcription of RNA was synthesized using a Transcriptor First Stand cDNA Synthesis Kit. Quantitative real-time PCR was performed using a LightCycler 480 Real-time PCR System (Roche) in accordance with the manufacturer’s instructions. The relative expression levels of the target genes were normalized against GAPDH gene expression, and the primers are shown in
Table 1
.
Table 1. The primers for real-time PCR
| Primer |
Sequence(5’ to 3’) |
|
ALK7-F
ALK7-R
|
GACATGAAAACATCCTTGGT
ACTTCTGGTCACAAACAACC
|
|
TNF-α-F
TNF-α-R
|
CAGCCTCTTCTCATTCCTGCT
GGGTCTGGGCCATAGAACTG
|
|
iNOS-F
iNOS-R
|
CATTCAGATCCCGAAACGCT
TGTAGGACAATCCACAACTCGC
|
|
COX2-F
COX2-R
|
AGAGGTGTATCCCCCCACAG
TGTCGCACACTCTGTTGTGC
|
|
IL-6-F
IL-6-R
|
TTGCCTTCTTGGGACTGATG
TCATTTCCACGATTTCCCAG
|
|
Arg-1-F
Arg-1-R
|
AAAGGCCGATTCACCTGAGC
AGGTAGTCAGTCCCTGGCTT
|
|
chi3I3-F
chi3I3-R
|
TGAAGGAGCCACTGAGGTCT
TGAAGGAGCCACTGAGGTCT
|
|
TGF-β-F
TGF-β-R
|
AGAGCCCTGGATACCAACTATTG
TGCGACCCACGTAGTAGACG
|
|
Mrc-1-F
Mrc-1-R
|
CGTTTCGGTGGACTGTGGA
GTTGTGGGCTCTGGTGGG
|
|
KLF4-F
KLF4-R
|
ACTAACCGTTGGCGTGAGGA
TTACTGCTGCAAGCTGCACC
|
|
SR-A-F
SR-A-R
|
TGGAGGAGAGAATCGAAAGCA
CTGGACTGACGAAATCAAGGAA
|
|
CD36-F
CD36-R
|
GACTGGGACCATTGGTGATGA
AAGGCCATCTCTACCATGCC
|
|
ABCA1-F
ABCA1-R
|
AGGCACTCAAGCCACTGCTTGT
TGCCTCTGCTGTCTAACAGCGT
|
|
ABCG1-F
ABCG1-R
|
GGTTGCGACATTTGTGGGTC
TTCTCGGTCCAAGCCGTAGA
|
|
PPARγ-F
PPARγ-R
|
GCTTGTGAAGGATGCAAGGG
GATATCACTGGAGATCTCCGCC
|
|
PPARδ-F
PPARδ-R
|
CGAGTTCTTGCGAAGTCTCC
CCGTCTTCTTTAGCCACTGC
|
|
IRF4-F
IRF4-R
|
GGTGAGGAGTTTCCAGACCC
GGTGAGGAGTTTCCAGACCC
|
|
NR4A-F
NR4A-R
|
GCTGCAGAATGACTCCACC
ACAGCAGCACTGGGCTTA
|
|
Nrf2-F
Nrf2-R
|
TCTTGGAGTAAGTCGAGAAGTGT
GTTGAAACTGAGCGAAAAAGGC
|
|
STAT6-F
STAT6-R
|
CCTGGTCGGTTCAGATGCTTT
GTGCGGCAAGATGCTGTTTC
|
|
STAT3-F
STAT3-R
|
CAAGGGCTTCTCCTTCTGGG
CCTGGGTCAGCTTCAGGATG
|
|
GAPDH-F
GAPDH-R
|
TGAAGGGTGGAGCCAAAAG
AGTCTTCTGGGTGGCAGTGAT
|
Western Blotting Analysis
Cellular proteins were lysed in RIPA assay buffer. Protein concentrations were then quantified with a BCA protein assay kit. The 40 micrograms of protein were separated by via SDS-PAGE on a 5% or 10% acrylamide gel and transferred to a PVDF membrane (Millipore), which was subsequently blocked with milk and incubated with the appropriate primary antibody overnight at 4℃. After incubation with the appropriate secondary antibody for one hour at room temperature, relative expression was visualized using a Fluor Chem E imager (Protein Simple, Fluor Chem E). The specific protein expression was normalized against GAPDH expression, and the antibodies are shown in
Table 2
.
Table 2. Antibody for Immunoblot
| Primary Antibody |
Cat No |
Manufacturer |
Sources of species |
| ALK7 |
sc-374538 |
Santa Cruz Biotechnology |
Mouse |
| iNOS |
ab3523 |
Abcam |
Rabbit |
| IL-6 |
AF-406-NA |
R&D Systems |
Goat |
| Arg-1 |
610708 |
BD Biosciences |
Mouse |
| KLF4 |
ab151733 |
Abcam |
Rabbit |
| CD36 |
sc-7309 |
Santa Cruz Biotechnology |
Mouse |
| SR-A |
sc-166139 |
Santa Cruz Biotechnology |
Mouse |
| ABCA1 |
ab7360 |
Abcam |
Rabbit |
| ABCG1 |
ab244442 |
Abcam |
Rabbit |
| PPARγ |
16643-1-AP |
Proteintech |
Rabbit |
| GAPDH |
2118 |
Cell Signaling Technology |
Rabbit |
Immunofluorescence Staining
We performed immunofluorescent staining of BMDMs as described
19)
. After incubating with primary antibodies at 4℃ overnight, the slices went through rewarming at 37℃ for half an hour and were washed with PBS before incubation with appropriate fluorescence-labeled secondary antibody for one hour. Cell nuclei were stained with DAPI, and images were captured using a confocal laser scanning microscope. The antibodies are shown in
Table 3
.
Table 3. Antibody for Immunofluorescence
| Primary Antibody |
Cat No |
Manufacturer |
Sources of species |
| ALK7 |
sc-374538 |
Santa Cruz Biotechnology |
Mouse |
| PPARγ |
16643-1-AP |
Proteintech |
Rabbit |
| CD68 |
Ab125212 |
Abcam |
Rabbit |
Statistical Analysis
All data were represented as the means±SD. Comparisons between groups were evaluated using a two-tailed Student t test or one-way ANOVAs. All statistical analyses were completed using SPSS, version 22.0. A value of P<0.05 were considered statistically significant.
Result
The Up-Regulation of ALK7 in Activated Macrophages
To determine ALK7’s potential role in macrophage activation in atherogenesis, isolated BMDMs and MPMs were treated with Ox-LDL. Notably, ALK7 mRNA was markedly upregulated upon Ox-LDL stimulation, as determined by RT-PCR (
Fig.1A)
, whereas minimal baseline ALK7 expression was observed with PBS treatment. The differential ALK7 mRNA expression was further confirmed at the protein level by western blot analysis (
Fig.1B)
. Moreover, double immunofluorescence staining revealed ALK7’s strong immunoreactivity in activated BMDMs, as evaluated by the increased ratio of ALK7(+)CD68(+) to ALK7(+)CD68(-) with Ox-LDL treatment compared to that of PBS administration (
Fig.1C)
. Collectively, these findings indicate that increased ALK7 expression was positively correlated with macrophage activation.
AKL7 Knockdown Attenuates Pro-Inflammatory Macrophage Polarization
Macrophages are responsible for the inflammatory response in atherogenesis, exhibit remarkable plasticity, and switch between classical activation (M1) and alternative activation (M2) phenotypes
7,
8)
. To explore upregulated ALK7’s contribution to macrophage activation, BMDMs were transfected with adenovirus harboring ALK7 short hairpin RNA (AdshALK7). The results showed that ALK7 expression decreased significantly in BMDMs after transfection with AdshALK7 (
Fig.2A)
. After administration of Ox-LDL, RT-PCR analysis revealed that the mRNA expression levels of marker genes related to the pro-inflammatory M1 macrophage phenotype (including TNF-α, iNOS, Cox-2, and IL-6) had decreased dramatically, and the mRNA expression levels of genes related to the anti-inflammatory M2 macrophage phenotype (including Arg-1, TGF-β, Mrc-1, and KLF4) had increased because of the ALK7 knockdown (
Fig.2B)
. Western blot analysis confirmed the changes in iNOS and IL-6 expression, as well as Arg-1 and KLF4 expression (
Fig.2C)
. Collectively, these data confirmed that ALK7 knockdown promoted the alternative activation of macrophages.

Accumulation of cholesterol in macrophages, and subsequent foam cell formation, is a critical initial step in macrophage activation and is implicated in atherogenesis
22)
. Neutral lipid staining with oil red O showed significantly decreased foam cell formation in BMDMs transfected with AdshALK7 (
Fig.3A)
. Taking into account that variations in cholesterol uptake and efflux can contribute to cholesterol accumulation and foam cell formation, the related markers for each process were tested
23)
. We observed that the mRNA levels of genes involved in cholesterol influx (CD36 and SR-A) decreased, whereas the mRNA levels of genes implicated in cholesterol efflux (ABCA1 and ABCG1) increased in BMDMs transfected with AdshALK7 (
Fig.3B)
. Additionally, the above results were further verified by Western blotting (
Fig.3C)
.
PPARγ is Required for the Effect of ALK7 Knockdown on Macrophage Inactivation
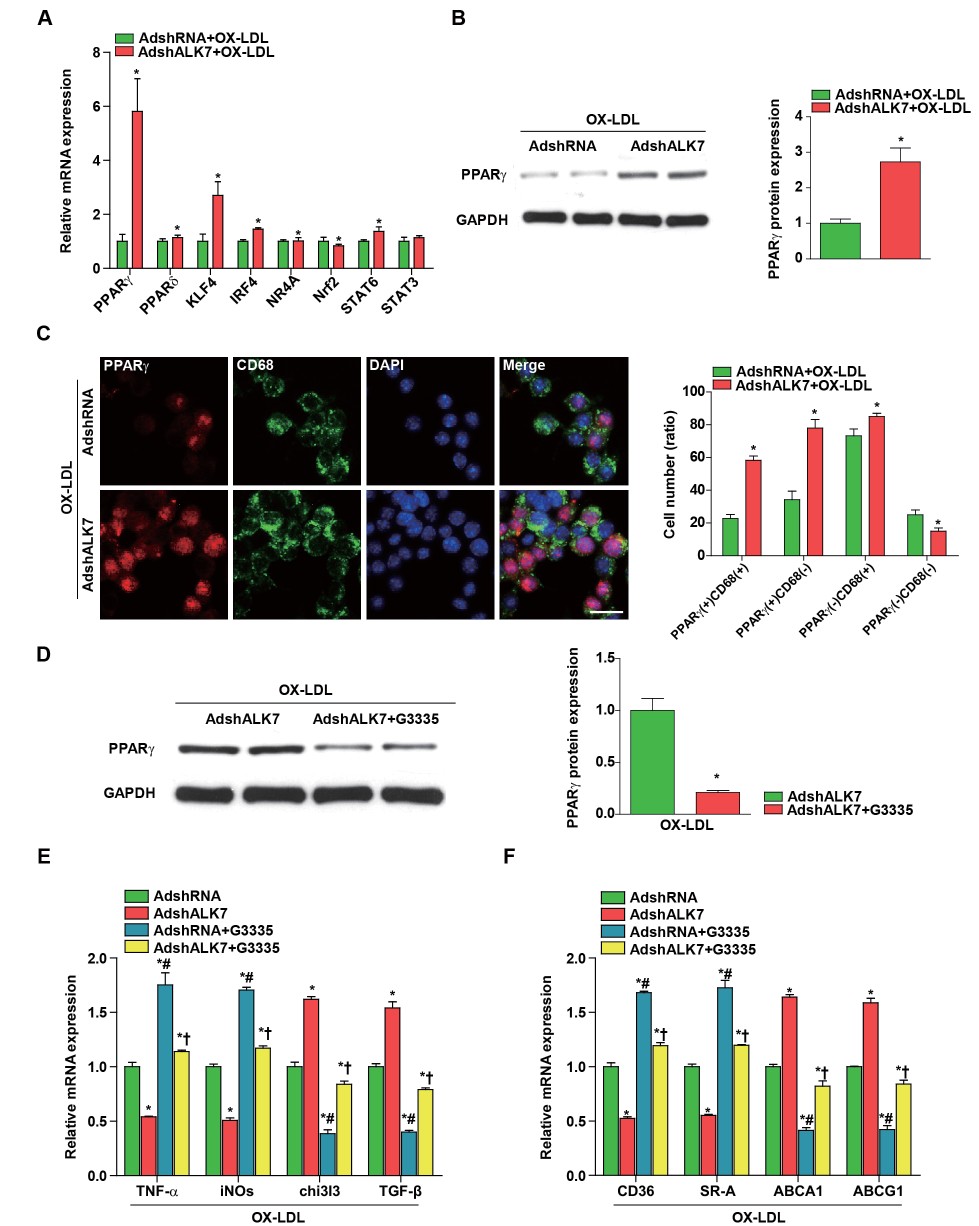
Next, ALK7’s evident macrophage activation prompted us to investigate the underlying mechanism. Previous studies have suggested that multiple targets are implicated in regulating macrophage polarization and atherogenesis, including PPARγ, PPARδ, KLF4, IRF4, NR4A, Nrf2, STAT6 and STAT3
24,
25)
. Among the genes tested in our study, PPARγ showed the greatest increase in mRNA in response to ALK7 silencing under stimulation with Ox-LDL (
Fig.4A)
. Western blot analysis further confirmed the up-regulation of PPARγ expression in BMDMs transfected with AdshALK7 (
Fig.4B)
. Using confocal microscopy, we further noticed dramatically increased PPARγ expression in the nuclei of macrophages with ALK7 knockdown, which was indicated by the increase in PPARγ positive cells [PPARγ(+)CD68(+) and PPARγ(+)CD68(-)] compared with those of the non-silenced groups (
Fig.4C)
. Additionally, a previous study indicated that PPARγ is responsible for the ALK7 silencing-mediated decrease in inflammatory adipocytokine secretion, which improved glucose tolerance and insulin sensitivity
16,
18)
. Subsequently, we examined whether PPARγ also mediated ALK7’s effect on macrophage activation in vitro. PPARγ expression was dramatically down-regulated in macrophages upon administration of a PPARγ-specific antagonist (G3335) (
Fig.4D)
. Functionally, PPARγ inactivation blunted ALK7 knockdown-mediated down-regulation of pro-inflammatory M1 phenotype cytokine expression (TNF-α and iNOs) and upregulation of anti-inflammatory M2 phenotype cytokine expression (chi3I3 and TGF-β) (
Fig.4E)
. Moreover, the decrease in expression of genes involved in cholesterol influx (CD36 and SR-A) and increase in expression of genes implicated in cholesterol efflux (ABCA1 and ABCG1) were largely reversed by PPARγ inactivation in the presence of Ox-LDL (
Fig.4F)
. Based on these findings, we concluded that PPARγ inactivation was required for ALK7-mediated macrophage activation.
Discussion
Macrophage activation is responsible for the development of atherosclerosis, which is the determinate inflammatory process underlying severe cardiovascular disease
6)
. Our current study first demonstrated that ALK7 was a novel independent indicator of atherogenesis, as exhibited by upregulated macrophages’ expression upon OX-LDL administration. By performing loss-of-function analysis of ALK7 in vitro, we observed that ALK7 knockdown promoted M2 phenotype macrophages but attenuated M1 macrophage switching. Furthermore, ALK7 knockdown significantly ameliorated foam cell formation by up-regulating ABCA1 and ABCG1, which are involved in cholesterol efflux, and down-regulating CD36 and SR-A, which are implicated in cholesterol influx. Mechanistically, silencing ALK7 dramatically promoted PPARγ expression, and the ALK7 knockdown-mediated inhibition of macrophage activation was largely reversed by treatment with G3335, a PPARγ inhibitor. Based on the present study’s results, we propose ALK7 has an unexplored role in regulating macrophage activation during atherosclerosis development, at least partially through regulating PPARγexpression
In recent decades, tremendous efforts have been made to elucidate the underlying mechanisms of atherosclerosis that effectively suppress development of atherogenesis-related cardiovascular diseases. Recently, TGF-β family members have been reported be upregulated in mouse and human atherosclerotic lesions
26)
. More importantly, many lines of evidence have demonstrated that TGF-β and activin A play pivotal and protective roles against the development of atherosclerotic lesions by modulating key cellular signals
27-
29)
. Notably, ALK7 is well recognized as an important receptor of the TGF-β family members, nodal, and activing
11)
. Consistent with the above findings, ALK7 may act as a novel target for atherosclerosis management. In agreement with our expectation, we found that ALK7 expression increased, primarily in macrophages upon OX-LDL administration using co-immunofluorescence staining of macrophage-specific markers. Previous studies indicated that activin A affected macrophage polarization through binding to the type 1 receptors ALK4 or ALK7, while M2-related macrophages can be induced by IL-4/13, IL-10, and TGF-β, which promote inflammation resolution under physiological conditions
30)
. As expected, by knocking down ALK7 in macrophages, we observed promotion of M2 polarization of macrophages but attenuation of M1 macrophage switching. Additionally, ALK7 silencing also affected macrophage-derived foam cell formation through increased expression of cholesterol efflux-associated ABCA1 and ABCG1 and decreased expression of cholesterol uptake-associated CD36 and SR-A.
There has been an impressive increase in the appreciation of inflammation’s importance in atherogenesis in recent decades. The most attention has been focused on the role of recruited macrophages in regulating the inflammatory response and foam cell formation within atherosclerotic lesions
9)
. Within this heterogeneous group of macrophages, there are classically activated, pro-inflammatory, M1 macrophages, and reparative M2 macrophages, which are derived by different pathways and have different targets
6,
25)
. Our study results demonstrate that PPARγ was the most significantly upregulated gene in macrophages, and this observation was verified by western blotting and co-immunofluorescence staining. Recent work has identified the nuclear hormone receptor PPARγ as a critical signaling molecule in determining macrophage phenotype in vitro and in atherosclerotic plaques
31)
. Specifically, PPARγ activation polarizes monocytes to become M2 macrophages, and M2 macrophages induced by PPARγ inhibit the secretion of pro-inflammatory mediators in M1 macrophages
32)
. Moreover, PPARγ is responsible for suppressing lipolysis that leads to fat accumulation in obesity
16)
. PPARγ is also known to decrease the secretion of inflammatory adipocytokines, thereby improving glucose tolerance and insulin sensitivity through ALK7 inactivation
18)
. Based on these observations, we hypothesized that PPARγ in macrophages is required for macrophage inactivation by ALK7 silencing. In the current study, we noticed that ALK7 knockdown significantly promoted M2 polarized macrophages but attenuated M1 polarized macrophages, while these effects were reversed by PPARγ inactivation.
Macrophages’ uptake of modified LDL in the subendothelium, and the subsequent transformation into foam cells, has been suggested to play a critical role in atherosclerosis development, which is regulated by an imbalance in cholesterol efflux, influx, and synthesis
9)
. PPARγ has emerged as an important regulator of foam cell formation via multiple signaling pathways
23)
. PPARγ activation represses SR-A and inhibits triglyceride accumulation via down-regulated apoB-48 receptor expression in human macrophages
33)
. PPARγ reduces ACAT1 mRNA levels to decrease the cholesterol esterification rate
34)
. Moreover, the positive regulation of cholesterol efflux in macrophages by PPARγ is highlighted by enhanced SR-B1, ABCA1, ABCG1, and apoE expression
35)
. In our study, we demonstrated that PPARγ activation was responsible for the ALK7 knockdown’s suppressive effect on foam cell formation through upregulation of ABCA1 and ABCG1 and down-regulation of CD36 and SR-A.
In conclusion, our findings demonstrated that silencing ALK7 expression in macrophages attenuated macrophage activation, as characterized by an increased ratio of M2 to M1 macrophages, as well as upregulation of the genes involved in cholesterol efflux and down-regulation of the genes involved in cholesterol influx. The protective effects of ALK7 knockdown against macrophage activation were partially mediated by up-regulating PPARγ expression. Our work highlights the important role of neutralizing ALK7 as a potential therapeutic approach for atherosclerosis management.
Sources of Funding
This work was supported by grants from the National Natural Science Foundation of China (81900266); The Fundamental Research Funds for the Central Universities (2042019kf0153); Zhongnan Hospital of Wuhan University Science, Technology and Innovation Seed Fund (znpy2018013); The project of Health commission of Hubei province (WJ2019M208).
Conflict of Interest
None.
References
- 1) Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation, 2017; 135: e146-e603
- 2) Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol, 2005; 25: 2255-2264
- 3) Ley K, Laudanna C, Cybulsky MI and Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol, 2007; 7: 678-689
- 4) Libby P. Inflammation in atherosclerosis. Nature, 2002; 420: 868-874
- 5) Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C and Li J. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signalling pathways. Cell Signal, 2014; 26: 192-197
- 6) Tabas I and Bornfeldt KE. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ Res, 2016; 118: 653-667
- 7) Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN and Wynn TA. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity, 2014; 41: 14-20
- 8) Mills CD. Anatomy of a discovery: m1 and m2 macrophages. Front Immunol, 2015; 6: 212
- 9) Tall AR and Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol, 2015; 15: 104-116
- 10) Pepper MS. Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev, 1997; 8: 21-43
- 11) Morikawa M, Derynck R and Miyazono K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb Perspect Biol, 2016; 8
- 12) Namwanje M and Brown CW. Activins and Inhibins: Roles in Development, Physiology, and Disease. Cold Spring Harb Perspect Biol, 2016; 8
- 13) Michael IP, Saghafinia S, Tichet M, Zangger N, Marinoni I, Perren A and Hanahan D. ALK7 Signaling Manifests a Homeostatic Tissue Barrier That Is Abrogated during Tumorigenesis and Metastasis. Dev Cell, 2019; 49: 409-424 e6
- 14) Li X and Ventura A. ALK7 Erects a Suppressive Barrier to Tumor Progression and Metastasis. Dev Cell, 2019; 49: 304-305
- 15) Kogame M, Matsuo S, Nakatani M, Kurisaki A, Nishitani H, Tsuchida K and Sugino H. ALK7 is a novel marker for adipocyte differentiation. J Med Invest, 2006; 53: 238-245
- 16) Yogosawa S, Mizutani S, Ogawa Y and Izumi T. Activin receptor-like kinase 7 suppresses lipolysis to accumulate fat in obesity through downregulation of peroxisome proliferator-activated receptor gamma and C/EBPalpha. Diabetes, 2013; 62: 115-123
- 17) Balkow A, Jagow J, Haas B, Siegel F, Kilic A and Pfeifer A. A novel crosstalk between Alk7 and cGMP signalling differentially regulates brown adipocyte function. Mol Metab, 2015; 4: 576-583
- 18) Carlsson LM, Jacobson P, Walley A, Froguel P, Sjostrom L, Svensson PA and Sjoholm K. ALK7 expression is specific for adipose tissue, reduced in obesity and correlates to factors implicated in metabolic disease. Biochem Biophys Res Commun, 2009; 382: 309-314
- 19) Huang H, Tang Y, Wu G, Mei Y, Liu W, Liu X, Wan N, Liu Y and Huang C. ALK7 protects against pathological cardiac hypertrophy in mice. Cardiovasc Res, 2015; 108: 50-61
- 20) Watanabe R, Yamada Y, Ihara Y, Someya Y, Kubota A, Kagimoto S, Kuroe A, Iwakura T, Shen ZP, Inada A, Adachi T, Ban N, Miyawaki K, Sunaga Y, Tsuda K and Seino Y. The MH1 domains of smad2 and smad3 are involved in the regulation of the ALK7 signals. Biochem Biophys Res Commun, 1999; 254: 707-712
- 21) Wang H and Tsang BK. Nodal signalling and apoptosis. Reproduction, 2007; 133: 847-853
- 22) Libby P, Ridker PM and Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature, 2011; 473: 317-325
- 23) Tall AR, Yvan-Charvet L, Terasaka N, Pagler T and Wang N. HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab, 2008; 7: 365-375
- 24) Sica A and Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest, 2012; 122: 787-795
- 25) Leitinger N and Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler Thromb Vasc Biol, 2013; 33: 1120-1126
- 26) Grainger DJ, Kemp PR, Metcalfe JC, Liu AC, Lawn RM, Williams NR, Grace AA, Schofield PM and Chauhan A. The serum concentration of active transforming growth factor-beta is severely depressed in advanced atherosclerosis. Nat Med, 1995; 1: 74-79
- 27) Hansson GK. Inflammatory mechanisms in atherosclerosis. J Thromb Haemost, 2009; 7 Suppl 1: 328-331
- 28) Molloy CJ, Taylor DS and Pawlowski JE. Novel cardiovascular actions of the activins. J Endocrinol, 1999; 161: 179-185
- 29) Kozaki K and Ouchi Y. Activin/follistatin and atherosclerosis--a review. J Atheroscler Thromb, 1998; 5: 36-40
- 30) Sierra-Filardi E, Puig-Kroger A, Blanco FJ, Nieto C, Bragado R, Palomero MI, Bernabeu C, Vega MA and Corbi AL. Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood, 2011; 117: 5092-5101
- 31) Duval C, Chinetti G, Trottein F, Fruchart JC and Staels B. The role of PPARs in atherosclerosis. Trends Mol Med, 2002; 8: 422-430
- 32) Charo IF. Macrophage polarization and insulin resistance: PPARgamma in control. Cell Metab, 2007; 6: 96-98
- 33) Moore KJ, Rosen ED, Fitzgerald ML, Randow F, Andersson LP, Altshuler D, Milstone DS, Mortensen RM, Spiegelman BM and Freeman MW. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat Med, 2001; 7: 41-47
- 34) Hirakata M, Tozawa R, Imura Y and Sugiyama Y. Comparison of the effects of pioglitazone and rosiglitazone on macrophage foam cell formation. Biochem Biophys Res Commun, 2004; 323: 782-788
- 35) Chinetti G, Gbaguidi FG, Griglio S, Mallat Z, Antonucci M, Poulain P, Chapman J, Fruchart JC, Tedgui A, Najib-Fruchart J and Staels B. CLA-1/SR-BI is expressed in atherosclerotic lesion macrophages and regulated by activators of peroxisome proliferator-activated receptors. Circulation, 2000; 101: 2411-2417


