Introduction
Familial hypercholesterolemia (FH) is an inherited disorder of lipoprotein metabolism caused by mutations of the genes involved in the LDL receptor-mediated pathway for cellular uptake of LDL. Most FH patients show an autosomal dominant trait. Hyper-LDL-cholesterolemia remains throughout their lives and causes premature coronary heart disease unless properly treated
1-
3)
.
Early diagnosis and initiation of lipid-lowering therapy is essential for preventing development of cardiovascular complications, even in Japan where cardiovascular disease is not the leading cause of death and its prevalence among FH patients seems to be somewhat lower than in Western countries. Heterozygous FH (HeFH) patients who carry the mutated gene in a single allele have plasma LDL cholesterol (LDL-C) levels double normal or higher and may experience the first cardiovascular event as early as their thirties. With mutations in both alleles, homozygous FH (HoFH) exhibits LDL-C levels twice those of HeFH, or even higher, and patients develop cardiovascular complications even in the first decade of their lives. Most HoFH is refractory to statins and other standard lipid-lowering drugs as most of them depend on remaining LDL receptor activity. Thus, HoFH has a poor prognosis compared to HeFH and therefore, earlier diagnosis and more aggressive treatments are required to prevent premature death.
1. Clinical Manifestations of HoFH
Familial hypercholesterolemia (FH) is a disease with a triad of clinical characteristics: elevated LDL-C, cutaneous and/or tendon xanthomas, and premature atherosclerotic cardiovascular disease (ASCVD). A genetic defect in LDL receptor function is the cause of FH, and activity of the LDL receptor is completely or almost completely absent in HoFH patients. HoFH includes homozygotes or compound/double heterozygotes of autosomal-dominant disease-causing mutations in the related genes, and their parents are HeFH, but there are rare exceptions that exhibit autosomal recessive inheritance
4,
5)
.
1) Elevated LDL-C Levels
HoFH patients have very high LDL-C levels from birth, which puts them at very high-risk of coronary heart disease. Plasma LDL-C levels are more than 500 mg/dL in many cases of genetically confirmed HoFH, but there is considerable variation in lipid levels among patients. Those with LDL-C levels over 370 mg/dL (or total cholesterol levels over 450 mg/dL) in the fasting steady state should consult with specialists as they are probably cases of HoFH.
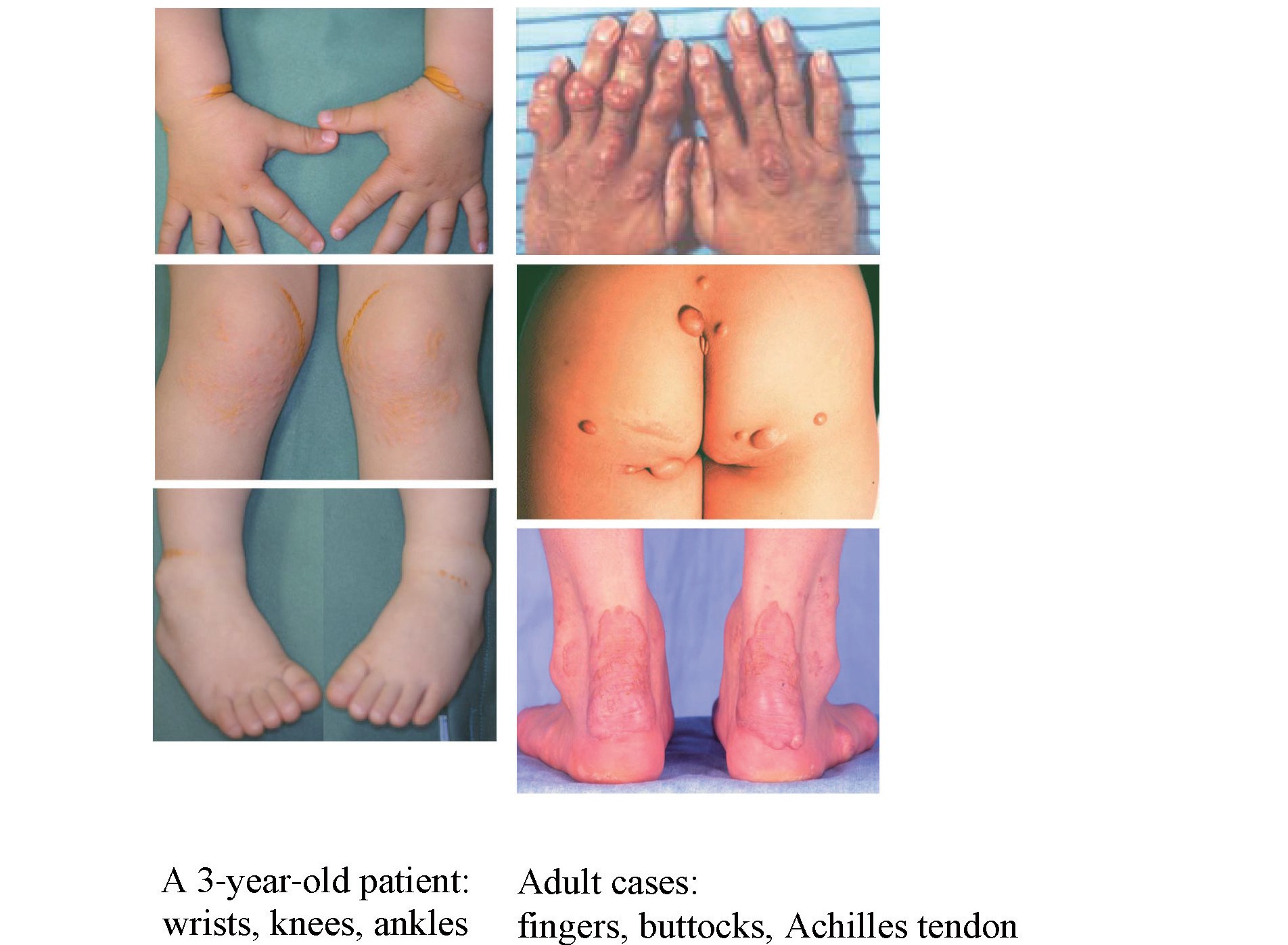
2) Cutaneous and Tendinous Xanthomas (
Fig.1
)
Characteristic cutaneous xanthomas that have developed since infancy are physical findings suggestive of HoFH. They are commonly found on the extensor surfaces of the elbows/knees and wrist/gluteal regions and parents often take a child to a doctor for the first visit because of them
6)
. Tendon xanthomas are pathognomonic for both HeFH and HoFH, but not apparent during childhood and gradually appear around puberty. Xanthomas can be repressed or made to regress with continuous aggressive lipid-lowering treatment.
3) ASCVD
The prognosis for untreated HoFH is extremely poor. It is difficult for patients to live beyond 30 years without treatment. The LDL-C accumulation threshold hypothesis, which uses a calculation of [LDL-C x years of life], has been proposed as a rational explanation for the coronary risk of FH, and according to it, the coronary threshold of HoFH would be around 11 years old even for individuals with lower LDL-C levels
7)
. Angina or myocardial infarction from infancy, as well as aortic supravalvular and/or valvular stenosis are often noted in HoFH
(
Fig.2)
, and may become the main cause of death in patients. Even with substantially effective treatment, systemic atherosclerosis, with such manifestations as aortic aneurysms, peripheral artery disease, and cerebrovascular disease, develops along with aging. Aggressive LDL-lowering treatments should be started as young as possible to prevent these atherosclerotic complications.
2. Prevalence
In the 1970s, prevalence of HeFH was estimated as 1 in 500 in the general population, and accordingly, that of HoFH as 1 in 1,000,000
8)
. However, more recent molecular genetic studies have revealed that FH is more common than previously expected. HeFH is now estimated as 1 in 200 to 300 and HoFH as 1 in 170,000 to 300,000 in many countries including Japan
9-
13)
.
3. Genetics
LDL receptor-related disease-causing mutations are identified only in 60 to 80% of clinically diagnosed HeFH
14-
16)
. Most of them are in the LDL receptor gene (
LDLR), and those in apolipoprotein B-100 (coded as
APOB gene), the main ligand for the LDL receptor, have also been reported mainly in Caucasian populations
17)
. In 2003, gain-of-function mutations in
PCSK9, the gene coding proprotein convertase subtilistin/kexin-type 9 and enhancer of LDL receptor degradation, were found. This was considered to be the second major FH-related gene, explaining 5% of HeFH in Japan
18)
.
Families with FH caused by mutations in
LDLR,
APOB, and
PCSK9 all show autosomal dominant inheritance. Since
LDLR accounts for the majority of the FH mutations, most individuals with HoFH are “true homozygotes” or “compound heterozygotes” for two different mutations in
LDLR. Some patients are “double heterozygotes”, having combined mutations in
LDLR and
PCSK9
4)
, for example. The parents of HoFH children who are “true or compound heterozygotes” mostly show the HeFH phenotype. On the other hand, the family traits of HoFH “double heterozygotes” may not apparently exhibit simple Mendelian inheritance since the heterozygotes for PCSK9 show a variety of phenotypes.
It should be noted that HeFH is underdiagnosed and undertreated in the general population
3)
. In the background of one suspected case of HoFH, there are many family members with HeFH , and they must be properly cared for in time to prevent their premature death.
Besides the mutations discussed above, those causing, autosomal recessive hypercholesterolemia (ARH), a rare unique type of FH have been reported in Japan
4,
5)
. ARH is caused by mutations in a gene coding low-density lipoprotein receptor adaptor protein 1 (
LDLRAP1). LDLRAP1 is an adaptor protein making a complex with clathrin and LDL receptors for efficient endocytosis of LDL receptors. Heterozygous carriers of this mutation do not exhibit the HeFH phenotype so it apparently shows autosomal recessive inheritance. If the parents in a probable case of HoFH have normolipidemia, ARH may be considered.
Disease-causing mutations cannot be identified in 20% to 40% of clinically diagnosed HeFH
19)
. This may be due to still-unknown FH-related genes or limitations of analytic technologies. A clinical diagnosis of typical HoFH cannot be excluded even if two pathogenic FH gene variants are not identified. A substantial portion of HeFH cases without “known” FH-gene mutations could be “polygenic hypercholesterolemia”
20)
or “oligogenic FH”
21-
23)
, though these concepts of FH have not become established. Although genetic analysis provides a definitive diagnosis of HoFH, the possibility of such cases cannot be excluded. Therefore, clinical assessment including thorough physical examination and familial studies is essential. For information, Japanese public healthcare insurance does not cover the expenses for HoFH genetic testing as of February 2021.
4. Pathophysiology
LDL receptor activity is completely or nearly all lost in HoFH. Severely elevated LDL-C levels from birth (more exactly, from fetal period
24)
) often cause fatal cardiovascular disease even in infancy. As an example of cholesterol deposition in tissues, pathognomonic skin xanthomas develop from infancy in HoFH and tendon xanthomas become apparent later, and they are more prominent than those in HeFH.
5. Diagnostic Criteria in Japan
1) Clinical Diagnosis
A clinical diagnosis of HoFH can be made on the basis of skin or tendon xanthomas since infancy, and untreated LDL-C levels of twice those of HeFH or higher. Diagnostic criteria for FH in Japanese guidelines apply not only HeFH but also HoFH
1,
4)
(
Table 1,
Table 2)
. In cases with very high LDL-C and/or prominent xanthomas, HoFH should be suspected
25,
26)
.
Table 1.
Diagnostic criteria for FH in children
|
1.Elevated serum LDL cholesterol levels: untreated LDL-C level of ≥ 140 mg/dL (If total cholesterol level is ≥ 220 mg/dL, measure LDL-C level)
2. Family history of FH or premature CAD (within second-degree relatives)
|
|
・Secondary hyperlipidemia should be ruled out.
・If a patient meets both of the above-mentioned criteria, FH is diagnosed.
・As LDL-C levels fluctuate during growth due to dietary and hormonal influences, careful examination is required.
・Clinical symptoms and findings including angina, xanthomas, and corneal arcus are rare in heterozygous FH children. Therefore, family history of FH is important in making diagnosis
・Premature CAD is defined as occurrence of CAD in men <55 years old or in women <65 years old.
・Homozygous FH should be suspected if patient has xanthomas.
|
Table 2.
Diagnostic criteria for heterozygous FH in adults (15 years of age or older)
|
1. Hyper-LDL-cholesterolemia (an untreated LDL-C level of >= 180 mg/dL)
2. Tendon xanthoma (tendon xanthoma on the backs of the hands, elbows, knees, etc. or Achilles tendon hypertrophy) or xanthoma tuberosum
3. Family history of FH or premature CAD (within the patient's second-degree relatives)
|
|
・The diagnosis should be made after excluding secondary hyperlipidemia.
・If a patient meets two or more of the above-mentioned criteria, the condition should be diagnosed as FH. In cases of suspected FH, obtaining a diagnosis using genetic testing is desirable.
・Xanthoma palpebrarum is not included in xanthoma tuberosum.
・Achilles tendon hypertrophy is diagnosed if the Achilles tendon thickness is >= 9 mm on X- ray imaging.
・An LDL-C level of >= 250 mg/dL strongly suggests FH.
・If a patient is already receiving drug therapy, the lipid level that led to treatment should be used as the reference for diagnosis.
・Premature CAD is defined as the occurrence of CAD in men <55 years of age or women<65 years of age, respectively.
・If FH is diagnosed, it is preferable to also examine the patient’s family members.
|
Typical HoFH exhibits total cholesterol levels of more than 600 mg/dL in total
1)
, but there is considerable overlapping of levels between HoFH and severe HeFH
28)
. LDL-C levels over 370 mg/dL (or total cholesterol levels over 450 mg/dL) in the fasting steady state would be sufficient for a diagnosis of probable HoFH and the patient should be referred to specialists. In pediatric patients, plasma LDL-C levels may fluctuate and should be measured multiple times.
Measurement of LDL receptor activity in the fibroblasts of patients provides useful information in diagnosing HoFH, but is currently not routinely available at the laboratories of commercial or research facilities in Japan. Assaying LDL receptor activity in lymphocytes is feasible
29)
but less reliable than fibroblast measurements.
Skin xanthomas since infancy are pathognomonic for HoFH, and sometimes the chief complaint for the first consultation with doctors
4)
(
Fig.1)
. Skin xanthomas in pediatric HoFH are frequently found in flexures of the wrist and ankles, and also in other regions having mechanical stress. Tendon xanthomas are more prominent than in HeFH
27)
, but become apparent later than skin xanthomas.
2) Genetic Diagnosis
Genetic testing is recommended for suspected HoFH cases, but is not covered by Japanese public healthcare insurance as of February 2021. Genetic diagnosis indicates the potential efficacy of drug treatment and thereby therapeutic strategies
30,
31)
. Conversely, drug ineffectiveness (for example, refractory to PCSK9 inhibitors) may suggest the HoFH genotype so genetic tests should be considered in such cases
32)
.
The possibility of detecting an FH-causative mutation in HeFH is 60 to 80%. Therefore, diagnosis should be carefully made together with clinical examinations and detailed familial studies even in cases of suspected HoFH where the mutations detected are apparently in 0 alleles or only 1 allele, because a genetic test does not exclude the possibility of HoFH due to unknown gene mutations. Consultation with experienced specialists is required in such cases.
6. Screening/Follow-Up for ASCVD
Most HoFH patients may die of ASCVD before 30 years old if untreated
33,
34)
. Once HoFH is suspected, extensive examination for ASCVD should be carried out. In HoFH, the potentially fatal disorders of angina pectoris, myocardial infarction, and aortic supra-valvular and valvular stenosis could occur even in childhood
(
Fig.2)
. Aortic supra-valvular and valvular stenosis in HoFH is sometimes difficult to treat and lethal
35)
. If patients are left untreated until around 20 years old, ASCVD risk is extremely high. Heart and aortic disease proceeds and systemic atherosclerosis develops later in life.
Non-invasive tests such as cardiac ultrasonography, carotid ultrasonography, and electrocardiograms should be conducted first, and enhanced CT for the aorta and coronary artery and coronary angiography should be considered when necessary
36)
. Exercise stress tests should be carefully performed with consideration for patient safety.
7. Differential Diagnosis
A differential diagnosis should be made using high LDL-C levels and prominent xanthomas. LDL-C may be elevated to HoFH levels by secondary hypercholesterolemia such as hypothyroidism or nephrotic syndrome. Xanthomas may develop due to hypercholesterolemia in primary biliary cholangitis. Sitosterolemia, caused by the ATP-binding cassette transporter ABCG5/ABCG8 gene (
ABCG5/ABCG8) mutations
37)
, sometimes exhibits elevation of LDL-C and skin xanthomas comparable to HoFH during the suckling stage
38)
. Although LDL-C elevation and skin xanthomas subside after weaning from breastfeeding, elevated levels of plasma plant-sterols (including sitosterol) persist in these patients. There is also cerebrotendinous xanthomatosis (CTX), an autosomal recessive disease caused by sterol 27-hydroxylase gene (
CYP27A) mutations, which is characterized by prominent xanthomatosis in the tendon and brain and sometimes accompanied by central nervous symptoms (mental retardation, cognitive impairment, or motor ataxia)
39)
. CTX is clinically diagnosed by elevated plasma cholestanol levels. A differential diagnosis should be carefully made because these diseases require specific therapeutic approaches.
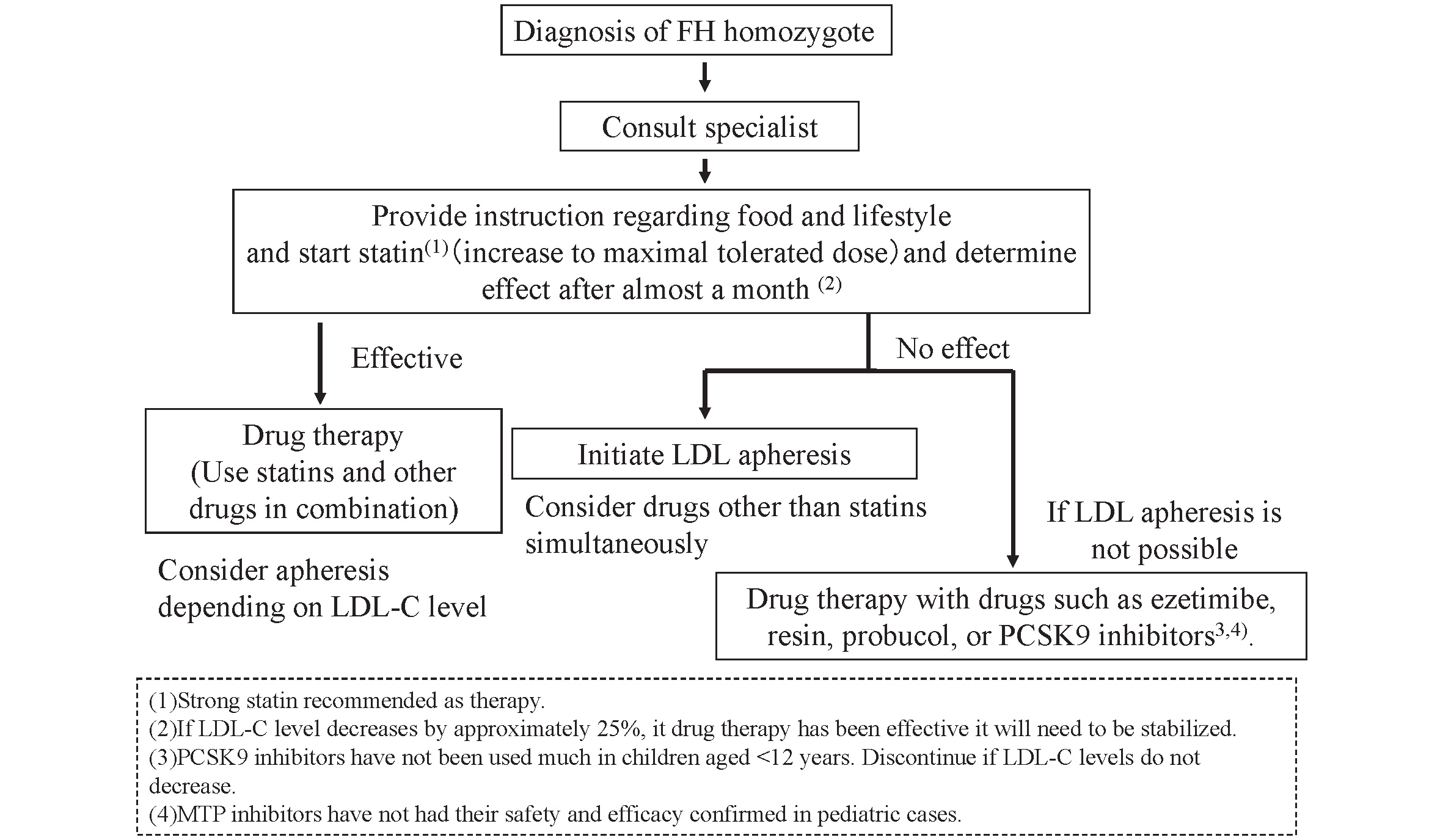
8. Treatment of HoFH
HoFH patients may suffer fatal ASCVD from infancy so initiation of aggressive LDL-C lowering as early as possible is essential for preventing their premature deaths
1,
4)
(
Fig.3,
Fig.4)
. Specific therapeutic strategies for preventing ASCVD development in individual cases should be planned in addition to LDL lowering.
Patients as well as their families will be burdened by various sources of stress such as anxiety about prognosis, concern about heredity, and costs of treatments. Supportive information should be provided, including that on possible financial aid, the therapeutic options available, and genetic counseling. Financial aid is available under The Program for Designated Intractable Diseases of the Japanese public healthcare system. Pediatric FH patients can receive support separately under The Program of Medical Aid for Chronic Pediatric Diseases of Specified Categories, which covers both HeFH and HoFH. In practice, consultation with specialists should strongly be advised.
1) Adult HoFH (15 years of age or older)
The LDL-C management goal is less than 100 mg/dL in primary prevention, and less than 70 mg/dL in secondary prevention
1)
in the Japanese guideline
(
Fig.3)
. Medication should start with statins at appropriate doses
40)
, followed by increasing them to maximal tolerated doses and combination with other drugs
41-
43)
. Achievement of these goals with drug treatment only is, however, not easy in HoFH.
Statins, ezetimibe and PCSK9 inhibitors all act by enhancing LDL receptor activity
44,
45)
, so their effectiveness depends on residual LDL receptor activity in the individual HoFH patient. Many HoFH patients are refractory to these drugs. However, if any of them are effective in lowering LDL-C, they should be continued together with the additional treatments described below.
MTP inhibitors (lomitapide) are a class of oral drug that is independent of LDL receptor activity. Clinically, they are indicated only for HoFH, and a decrease in LDL-C to approximately half of the pretreatment level can be achieved in many HoFH cases, if tolerated
46-
49)
. The main adverse effects are gastrointestinal symptoms and liver enzyme elevation accompanied by fat accumulation, similar to the symptoms observed in patients with MTP deficiency
50)
. The MTP inhibitor should be started with the lowest dose, taking adequate measures in accordance with nutritional guidance (restricted fat and alcohol intake), and then the dose should be carefully increased gradually.
Lipoprotein apheresis directly removes LDL from plasma through selective absorption or filtration of LDL using an extra-corporal circulation system, and this has been the core therapy for HoFH to date
51-
53)
. LDL-C levels can be precisely decreased by this procedure, depending upon the treated plasma volume, when properly performed. Lipoprotein apheresis not only removes LDL but also has potential pleiotropic effects in preventing atherosclerosis through removal of cell adhesion molecules
54)
, coagulation factors
55)
, and inflammatory cytokines
56)
. Lipoprotein apheresis for HoFH once every 1 to 2 weeks is covered by Japanese public healthcare insurance. The use of ACE inhibitors is contra-indicated for patients treated by lipoprotein apheresis with the Liposorber system and selective absorption of LDL by dextran sulfate-cellulose because shock may occur due to an increase in bradykinin activation.
Another option is liver transplantation. While this is highly invasive, it has been shown be a feasible therapeutic option for reversing atherosclerotic changes in HoFH patients that are uncontrollable with conservative therapy
57,
58)
.
2) Pediatric HoFH
Even in the suckling stage, children with HoFH must be referred to specialists for initiation of lipid-lowering therapy, as this is the key to a better prognosis
4)
. The target LDL-C level should be the same as for adult HoFH in secondary prevention (though not clearly stated in the current pediatric guideline
4)
). In pediatric HeFH, it is set at less than 140 mg/dL for primary prevention.
Statins and life-style interventions must be started at the time of diagnosis. Statins must be up-titrated and efficacy should be evaluated within a 1-month interval in order to titrate up to the maximal tolerated doses. Combination therapy with ezetimibe, bile sequestering resins, and PCSK9 inhibitors should be considered. These strategies may be effective in cases where there is a response in residual LDL receptor activity. Probucol should also be considered as it could reduce LDL in HoFH due to an unknown mechanism. Administration of an MTP inhibitor could be considered but only with extreme caution because no results of clinical trials in children have been reported. Drug therapy should be conducted until it becomes possible to commence lipoprotein apheresis.
In any case, lipoprotein apheresis must be considered since LDL-C targets are seldom achieved in HoFH only with drug treatment. This extracorporeal circulation therapy is generally commenced at the age of 5 or older, though it has reportedly been started in a patient who was 3.5 years old. In Japan, the main method is selective LDL absorption by dextran sulfate cellulose. However, with the aim of reducing extracorporeal volume, simple plasma exchange can be selected for children with a bodyweight under 30 kg.
3) Treatment during Pregnancy in HoFH
Female HoFH patients could be treated in the same way as for secondary prevention on reaching childbearing age. During pregnancy, LDL and VLDL are generally increased, and continuation of LDL-lowering therapy would be important. Statins and many other drugs are contraindicated during pregnancy and breastfeeding, and only bile sequestering resins can be used, taking proper care, but their LDL-lowering effects are limited
59)
. It has been reported that lipoprotein apheresis is effective and feasible during pregnancy, and a case of cardiovascular death caused by suspension of apheresis during pregnancy
60)
has been reported. Consultation with experienced specialists is recommended for pregnancy in HoFH.
Conflicts of Interest
Atsushi Nohara has nothing to disclose. Hayato Tada has nothing to disclose. Masatsune Ogura has received honoraria from Amgen Inc., Astellas Pharma Inc. Sachiko Okazaki has received scholarship grants from Minophagen Pharmaceutical Co., Ltd., Kowa Company, Ltd. Koh Ono has nothing to disclose. Hitoshi Shimano has nothing to disclose. Hiroyuki Daida has received honoraria from Amgen Inc., Daiichi-Sankyo Co., Ltd., Kowa Co., Ltd., and MSD K.K., Novartis Pharma K.K., Bayer Yakuhin, Ltd. and received clinical research funding from Canon Medical Systems Corporation, Philips Japan, Ltd., Toho Holdings Co., Ltd., Asahi Kasei Corporation, and Inter Reha Co., Ltd. HD has also received scholarship grants from Nippon Boehringer Ingelheim Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Sanofi K.K., MSD K.K., Daiichi-Sankyo Co., Ltd., Pfizer Co., Ltd., Mitsubishi Tanabe Pharma Corp., Astellas Pharma Inc., Takeda Pharmaceutical Co., Ltd., Teijin Pharma, Ltd., Shionogi & Co., Ltd., Actelion Pharmaceuticals, Ltd., Actelion Ltd., Kowa Co., Ltd., Bayer Yakuhin, Ltd. HD has also courses endowed by companies, including Philips Japan, Ltd., ResMed, Fukuda Denshi Co., Ltd., and Paramount Bed Co., Ltd. Kazushige Dobashi has nothing to disclose. Toshio Hayashi has nothing to disclose. Mika Hori has nothing to disclose. Kota Matsuki has nothing to disclose. Tetsuo Minamino has nothing to disclose. Shinji Yokoyama has nothing to disclose. Mariko Harada-Shiba has received stock holdings or options from Liid Pharma, honoraria from Amgen Inc., Astellas Pharma Inc., Sanofi, and scholarship grants from Aegerion Pharmaceuticals, Inc., Recordati Rare Diseases Japan, and Kaneka Corporation. Katsunori Ikewaki has nothing to disclose. Yasushi Ishigaki has nothing to disclose. Shun Ishibashi has received honoraria from Kowa Co., Ltd., and a scholarship grant from Ono Pharmaceutical Co., Ltd. Kyoko Inagaki has nothing to disclose. Hirotoshi Ohmura has nothing to disclose. Hiroaki Okazaki has received scholarship grants from Minophagen Pharmaceutical Co., Ltd., Kowa Company, Ltd. Masa-aki Kawashiri has nothing to disclose. Masayuki Kuroda has nothing to disclose. Masahiro Koseki has received clinical research funding from Kowa Company, Ltd., Rohto Pharmaceutical Co., Ltd. Takanari Gotoda has nothing to disclose. Shingo Koyama has nothing to disclose. Yoshiki Sekijima has nothing to disclose. Manabu Takahashi has nothing to disclose. Yasuo Takeuchi has nothing to disclose. Misa Takegami has nothing to disclose. Kazuhisa Tsukamoto has received honoraria from Bayer Yakuhin, Ltd., MSD Ltd., Takeda Pharmaceutical Company Ltd., and scholarship grants from Mitsubishi Tanabe Pharma Corporation., Bayer Yakuhin, Ltd., Sanofi K.K. Atsuko Nakatsuka has nothing to disclose. Kimitoshi Nakamura has nothing to disclose. Satoshi Hirayama has nothing to disclose. Hideaki Bujo has nothing to disclose. Daisaku Masuda has received clinical research funding from MSD K.K., Ono Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Kowa Co., Ltd. Takashi Miida has nothing to disclose. Yoshihiro Miyamoto has nothing to disclose. Takeyoshi Murano has nothing to disclose. Takashi Yamaguchi has nothing to disclose. Shizuya Yamashita has received honoraria from Kowa Company, Ltd., MSD K.K. Masashi Yamamoto has nothing to disclose. Koutaro Yokote has received honoraria from Kowa Company, Ltd., MSD K.K., Astellas Pharma Inc., Mitsubishi Tanabe Pharma Corp., Amgen K.K., Takeda Pharmaceutical Company Limited, Sanofi K.K., Ono Pharmaceutical Co., Ltd., AstraZeneca K.K., Daiichi-Sankyo Co., Ltd., Novartis Pharma K.K., Sumitomo Dainippon Pharma Co., Ltd., Kyowa Kirin Co., Ltd., Pfizer Japan Inc., Novo Nordisk Pharma Ltd., Nippon Boehringer Ingelheim Co., Ltd., Eli Lilly Japan K.K., Taisho Pharmaceutical Co., Ltd., Janssen Pharmaceutical K.K., and received clinical research funding from Taisho Pharmaceutical Co., Ltd. KY has also received scholarship grants from Mitsubishi Tanabe Pharma Corp., Takeda Pharmaceutical Co., Ltd., MSD K.K., Pfizer Japan Inc., Novo Nordisk Pharma Ltd., Taisho Pharmaceutical Co., Ltd., Kao Corporation, Ono Pharmaceutical Co., Ltd., Eli Lilly Japan K.K., Sumitomo Dainippon Pharma Co., Ltd., Nippon Boehringer Ingelheim Co., Ltd., Daiichi-Sankyo Co., Ltd., Teijin Pharma, Ltd., Shionogi Co., Ltd., Bayer Yakuhin, Ltd. Jun Wada has nothing to disclose.