Review
Role of Chronic Kidney Disease (CKD)–Mineral and Bone Disorder (MBD) in the Pathogenesis of Cardiovascular Disease in CKD
2023 Volume 30 Issue 8 Pages 835-850

Details
2023 Volume 30 Issue 8 Pages 835-850
Cardiovascular disease (CVD) is the leading cause of death in patients with chronic kidney disease (CKD). Multiple factors account for the increased incidence of cardiovascular morbidity and mortality in patients with CKD. Traditional risk factors for atherosclerosis and arteriosclerosis, including age, hypertension, dyslipidemia, diabetes mellitus, and smoking, are also risk factors for CKD. Non-traditional risk factors specific for CKD are also involved in CVD pathogenesis in patients with CKD. Recently, CKD–mineral and bone disorder (CKD–MBD) has emerged as a key player in CVD pathogenesis in the context of CKD. CKD–MBD manifests as hypocalcemia and hyperphosphatemia in the later stages of CKD; however, it initially develops much earlier in disease course. The initial step in CKD–MBD involves decreased phosphate excretion in the urine, followed by increased circulating concentrations of fibroblast growth factor 23 (FGF23) and parathyroid hormone (PTH), which increase urinary phosphate excretion. Simultaneously, the serum calcitriol concentration decreases as a result of FGF23 elevation. Importantly, FGF23 and PTH cause left ventricular hypertrophy, arrhythmia, and cardiovascular calcification. More recently, calciprotein particles, which are nanoparticles composed of calcium, phosphate, and fetuin-A, among other components, have been reported to cause inflammation, cardiovascular calcification, and other clinically relevant outcomes. CKD–MBD has become one of the critical therapeutic targets for the prevention of cardiovascular events and is another link between cardiology and nephrology. In this review, we describe the role of CKD–MBD in the pathogenesis of cardiovascular disorders and present the current treatment strategies for CKD–MBD.
Abbreviations:CKD, chronic kidney disease; CKD-MBD, chronic kidney disease-mineral and bone disorder; CPPs, calciprotein particles; CPP1, primary CPPs; CPP2, secondary CPPs; CVC, cardiovascular calcification; CVD, cardiovascular disease; FGF, fibroblast growth factor; FGF23, fibroblast growth factor 23; KDIGO, Kidney Disease: Improving Global Outcomes; PTH, parathyroid hormone; RCT, randomized controlled trial; VDRA, vitamin D receptor activator; VSMC, vascular smooth muscle cell
Cardiovascular (CVD) is the leading cause of death in patients with chronic kidney disease (CKD), especially those undergoing dialysis therapy1, 2). Importantly, both atherosclerosis and arteriosclerosis are accelerated in patients with kidney failure. In patients with CKD, traditional risk factors for atherosclerosis and arteriosclerosis, such as advanced age, hypertension, dyslipidemia, diabetes mellitus, obesity, and smoking, are present3, 4). Furthermore, increasing evidence shows that non-traditional risk factors specific for CKD play pivotal roles in CVD pathogenesis. These factors include renal anemia; iron deficiency; chronic inflammation; increased oxidative stress; retention of uremic toxins, such as indoxyl sulfate, P-cresyl sulfate, and trimethyl-monoamine-N-oxide; and CKD–mineral and bone disorder (CKD-MBD)1, 3). Among the non-traditional risk factors, CKD–MBD has emerged as an important player in CVD pathogenesis5-7). Mineral and bone disease in CKD, now referred to as CKD–MBD, was once defined as a disorder of only bone and the parathyroid glands, termed renal osteodystrophy. However, CKD–MBD is now regarded as a systemic disorder that affects multiple organs and systems and is associated with an increased risk of mortality through CVD, bone fracture, and other complications5-7). CKD–MBD develops early in CKD; therefore, detecting CKD–MBD at an early stage of CKD is extremely important. Monitoring serum CKD–MBD-related mediators and taking effective measures are also important to prevent the progression of CKD–MBD and CVD8, 9). In the present study, we review the pathophysiology of CKD–MBD, the impact of CKD–MBD on the cardiovascular system, and the potential treatment strategies for CKD–MBD in both dialysis-independent and dialysis-dependent CKD populations.
The kidneys play a central role in mineral and bone metabolism10, 11). Healthy kidneys are under neural and hormonal regulation and respond to neurohumoral input by minutely changing the concentrations of calcium and phosphate ions in the urine. Both calcium and phosphate are filtered through the glomerular capillaries in the kidneys and undergo resorption in the renal tubular cells to achieve calcium and phosphate balance. Bone metabolism is also involved in the tight regulation of calcium and phosphate metabolism12). Parathyroid hormone (PTH), a hormone secreted by the parathyroid glands, acts on bone cells and regulates calcium and phosphate release into the circulation. PTH synthesis and secretion are mainly regulated by the serum concentrations of calcium, phosphate, and calcitriol (1,25-dihydrovyvitamin D). PTH also acts on renal proximal tubular cells and downregulates phosphate transporters, such as type 2 sodium–phosphate transporters, in the renal proximal cells, thereby leading to phosphaturia13). PTH upregulates 1-α hydroxylase in the renal proximal tubules, resulting in increased calcitriol production, which enhances calcium absorption from the small intestine and renal distal tubules. As for phosphate homeostasis, PTH and fibroblast growth factor (FGF) 23 together serve as phosphatonins, which are hormones that elevate urinary phosphate excretion. FGF23 is a phosphaturic hormone that is secreted by bone cells, mainly osteocytes. The regulation of FGF23 synthesis, modification, and degradation is complex and mediated by various factors, including phosphate loading, PTH, iron deficiency, inflammation, and aldosterone14). However, phosphate overload is one of the strong drivers of an elevation in intact serum FGF23. FGF23 downregulates type II sodium-dependent phosphate transporters in the renal proximal tubular cells and deactivates calcitriol, in turn reducing the serum phosphate concentration. Notably, FGF23 requires α-klotho as a co-receptor for the FGF receptor15). α-Klotho is mainly expressed in the renal distal tubules, choroid plexus, and parathyroid glands. The physiological action of FGF23 is limited to the organs expressing the gene encoding α-klotho, although an α-klotho-independent action of FGF23 has been reported. Indeed, recent basic studies have shown that the soluble form of α-klotho, but not membrane-bound α-klotho, may act on remote organs and play diverse physiological roles16).
Although the kidneys play a central role in the regulation of mineral and bone metabolism, the kidneys form a complex mineral network in concert with the parathyroid glands, bone, and gastrointestinal tract in the human body. The simplified interactions among the main CKD–MBD mediators are summarized in Fig.1. The flow of calcium and phosphate in relation to the mechanisms of action of FGF23, PTH, calcitriol, α-klotho, and calciprotein particles (CPPs) is illustrated in Fig.2.

The arrows represent the actions that increase the associated CKD–MBD mediators, while the capped lines represent the actions that decrease the associated CKD–MBD mediators. The overall impact of changes in each CKD–MBD parameter depends on the medical conditions of each patient.
CKD–MBD, chronic kidney disease–mineral and bone disorder; CPP1, primary calciprotein particles; CPP2, secondary calciprotein particles; FGF23, fibroblast growth factor 23; PTH, parathyroid hormone.

Calcium and inorganic phosphate balance are maintained by orchestration of the complex function exerted by the kidneys, parathyroid glands, bone, and gastrointestinal tract. This orchestration is mainly mediated by PTH, FGF23, and calcitriol (1,25-dihydroxyvitamin D). PTH downregulates sodium–phosphate transporters in the renal proximal tubules and increases urinary inorganic phosphate excretion. PTH also increases the absorption of calcium in the renal distal tubules, upregulates 1-α hydroxylase, and elevates calcitriol. FGF23 increases urinary inorganic phosphate excretion by downregulating sodium–phosphate transporters in the renal proximal tubules, and deactivates calcitriol. Calcitriol increases calcium and inorganic phosphate absorption via the gastrointestinal tract and calcium reabsorption via the renal proximal tubules. Calcitriol also increases FGF23 synthesis and secretion in bone. Calcitriol and PTH affect bone turnover and regulate bone mass, followed by changes in the amount of calcium/inorganic phosphate release into the bloodstream. Excessive calcium and inorganic phosphate accelerate cardiovascular calcification.
Ca, calcium; CKD, chronic kidney disease; CKD–MBD, chronic kidney disease–mineral and bone disorder; CPP1, primary calciprotein particles; CPP2, secondary calciprotein particles; FGF23, fibroblast growth factor 23; Pi, inorganic phosphate; PTH, parathyroid hormone; PTG, parathyroid gland.
As kidney function declines, the number of nephrons decreases in line with the decreased expression of α-klotho in the renal distal tubules. As a result, the action of FGF23 in the kidney becomes blunted, leading to the accumulation of inorganic phosphate in the body17). To avoid relative phosphate overload, the remaining nephrons need to compensate and increase the amount of urinary phosphate excretion per nephron. To meet this demand, the body accelerates the secretion and synthesis of FGF23 and PTH in the bone and parathyroid glands, respectively18, 19). The pathophysiology of increased synthesis and secretion of PTH and histological hyperplasia of the parathyroid glands are historically termed uremic secondary hyperparathyroidism. The underlying mechanism is being revised after the discovery of FGF23 and its action of phosphatonins19). Importantly, an increase in circulating FGF23 precedes PTH elevation. Basic studies have shown that increased circulating FGF23 deactivates calcitriol by upregulating 25-hydroxyvitamin D–24-hydroxylase. Deactivation of calcitriol leads to increased PTH synthesis and secretion and a decrease in calcium and phosphate absorption from the gut. However, increased PTH stimulates calcitriol production and compensates for the decreased calcium absorption in the gut. FGF23 and PTH co-operatively downregulate type II sodium–phosphate transporters expressed in the renal proximal tubules, resulting in a decrease in phosphate resorption and an increase in urinary phosphate excretion20). Hence, the serum concentrations of calcium and phosphate are maintained within normal ranges in early CKD at the cost of a compensatory increase in serum FGF23 and PTH concentrations, and a resulting decrease in serum calcitriol. These underlying mechanisms to avoid overt hyperphosphatemia and hypocalcemia until late CKD are referred to as the “trade-off” hypothesis21). Unfortunately, as kidney function continues to decline, the aforementioned compensatory actions become ineffective22). Patients finally develop hyperphosphatemia and hypocalcemia in late-stage CKD.
Overall, in line with the decline in kidney function, the serum α-klotho concentration decreases and the serum FGF23 concentration rises. Then, the serum calcitriol concentration is suppressed, in turn increasing the serum PTH concentration. Finally, the serum phosphate concentration increases and the serum calcium concentration decreases in stages G4 and G5 CKD8-10). The typical chronological changes in serum CKD–MBD biomarkers in relation to the decline in glomerular filtration rate are illustrated in Fig.3.

CKD, chronic kidney disease; CKD–MBD, chronic kidney disease–mineral and bone disorder; FGF23, fibroblast growth factor 23; PTH, parathyroid hormone.
The term CKD–MBD was first introduced to the field of nephrology in exchange for the term renal osteodystrophy, which had long been used to express the concept of mineral and bone disorder in the uremic milieu. Now, the use of the term renal osteodystrophy is limited to indicate bone disorders in CKD–MBD5). The newly coined CKD–MBD is not limited to disorders in the parathyroid glands and bone; rather, it represents a systemic disorder that causes cardiovascular calcification (CVC), bone disorder, and other complications, all of which together increase the incidence of death6, 7). CKD–MBD comprises the following three components: laboratory abnormalities, bone disorders, and CVC. In the following passages, we introduce the concept of each component.
1) Laboratory Abnormalities: The First Component of CKD–MBDCKD–MBD is often recognized when patients exhibit abnormal serum mineral concentrations. In pre-dialysis CKD, high serum concentrations of FGF23, PTH, and phosphate, as well as decreased serum concentrations of calcitriol, calcium, and α-klotho, are commonly observed8, 9). In dialysis, hypocalcemia, hypercalcemia, hyperphosphatemia, and high serum concentrations of PTH and FGF23 are often observed. Observational studies have consistently shown that abnormalities in serum CKD–MBD mediators are associated with an increased risk of morbidity and mortality in pre-dialysis and dialysis-dependent CKD5-7). The recommended ranges for serum concentrations of calcium, phosphate, and PTH have been set in both pre-dialysis and dialysis-dependent CKD populations6, 23, 24).
2) Cardiovascular Calcification: The Second Component of CKD–MBDCVC is a component of CKD–MBD6). According to epidemiological studies, CKD patients with CVC are at an increased risk of cardiovascular death25). CVC is defined as the deposition of calcium–phosphate crystals in the vascular wall and cardiac valves. Arterial calcification is divided into two forms: arterial intimal calcification and medial calcification. Both forms have shared and distinct clinical impacts on the cardiovascular system. In CKD, valvular and arterial calcification are accelerated. CVC was once considered a passive deposition of calcium–phosphate crystals in the cardiovascular system. However, CVC is now regarded as an actively regulated and cell-mediated process that resembles bone formation26). Vascular smooth muscle cells (VSMCs), macrophages, endothelial cells, and vascular interstitial cells (VICs) are involved in the calcification process26, 27). CVC occurs on the extracellular matrix in the cardiovascular system. Historically, the most important cell-mediated calcification process was believed to be the transformation of mesenchymal cells, including VSMCs and VICs, from the contractile phenotype to the synthetic phenotype, which is often referred to as osteochondrogenic differentiation. Runx2, osterix, and Msx2 are key transcription factors that determine the phenotypic change into osteoblast-like cells28). Osteoblast-like cells in the vascular beds and cardiac valves synthesize and secrete bone-related proteins, including osteopontin, osteocalcin, and collagen, which collectively accelerate extracellular matrix calcification. In the past 20 years, basic studies have revealed that diverse cell-mediated processes are involved in the pathogenesis of CVC. Apoptosis plays a critical role in the progression of CVC. For example, apoptotic bodies serve as calcification nuclei and accelerate CVC29). Other mechanisms include degradation of the extracellular matrix, synthesis and secretion of abnormal matrix vesicles, increased oxidative stress, cellular senescence, mitochondrial dysfunction, endoplasmic reticulum and autophagy disorders, and acquisition of osteogenic microRNA profiles in the extracellular vesicles30-33). Another important aspect of CVC is the imbalance between calcification promoters and inhibitors. In CKD, calcification inducers, such as hyperphosphatemia, hypercalcemia, oxidative stress, inflammatory cytokines, and uremic toxins, are highly prevalent, whereas calcification inhibitors, including fetuin-A, albumin, and ionized magnesium, are decreased. Moreover, vitamin K-dependent anti-calcification proteins are dysfunctional. Together, CVC is a complex process in which multiple cell-mediated processes are involved. For the prevention of CVC, a multifaceted approach targeting each step of the CVC pathology may be mandatory34). The interrelationship between the risk factors and the pathomechanisms of CVC are illustrated in Fig.4.
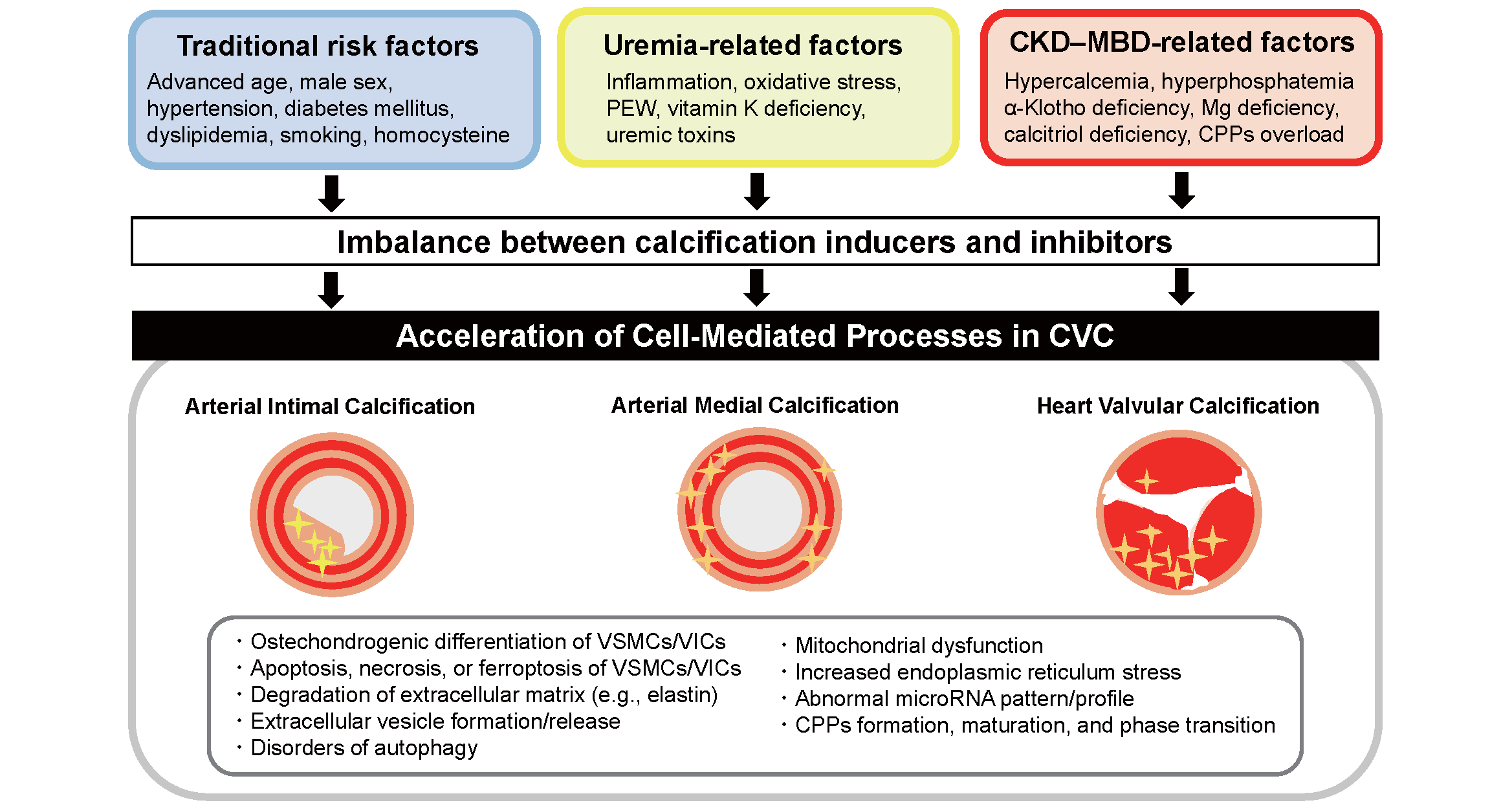
Patients with CKD are at an elevated risk of cardiovascular calcification, including arterial intimal and medial calcification and cardiac valvular calcification. The reasons for the heightened risk of CVC are supposed to be accumulation of multiple risk factors, including traditional risk factors for atherosclerosis, uremia-related risk factors, and CKD–MBD.
CKD, chronic kidney disease; CKD–MBD, chronic kidney disease–mineral and bone disorder; CPPs, calciprotein particles; CVC, cardiovascular calcification; Mg, magnesium; PEW, protein-energy wasting; VSMCs, vascular smooth muscle cells; VICs, valvular interstitial cells.
Very recently, CPPs have attracted more attention with respect to the pathogenesis of CVC35). CPPs are nanoparticles composed of calcium, phosphate, and serum proteins, including fetuin-A, albumin, and other acidic proteins. Because calcium and phosphate easily precipitate in the blood, fetuin-A and other proteins entrap and cover the calcium and phosphate ions and serve as a mineral chaperone to prevent hydroxyapatite formation in the circulation and physiologically transfer calcium and phosphate to bone and other tissues36). Calcium, phosphate, fetuin-A, and other proteins first form calcium–phosphate monomers, before maturing into primary CPP (CPP1) and finally evolving into secondary CPP (CPP2) in the circulation and tissue. CPP2 (but not CPP1) directly induces inflammation and accelerates phenotypic changes in VSMCs, resulting in calcification of the vascular wall and heart valves37). CPP2 also induces the synthesis of inflammatory cytokines in leukocytes, macrophages, VSMCs, and VICs38). Activation of inflammasomes in response to CPP2 is now regarded as one of the critical steps in CVC39).
3) Bone Fracture and Abnormal Bone Metabolism: The Third Component of CKD–MBDCKD–MBD involves bone abnormalities related to CKD5-7). Patients with CKD are at an increased risk of bone fracture40, 41). Bone fracture often develops in patients with decreased bone strength when exposed to mechanical forces, such as falls. Patients with CKD are at an elevated risk of osteoporosis and sarcopenia42, 43), and sarcopenia is a strong risk factor for falls and bone fracture44). Uremic secondary hyperparathyroidism, which is characterized by high circulating PTH concentrations and parathyroid hyperplasia in CKD, induces high turnover of bone and histologically presents as osteitis fibrosa, which is the most advanced bone phenotype in CKD45). In patients with advanced uremic secondary hyperparathyroidism, bone is often fragile, leading to bone fracture. Osteomalacia and adynamic bone status are also observed in some patients with CKD. Patients who develop bone fracture are at an increased risk of decreased activities of daily living, infection, bedsores, and death46). Accordingly, controlling secondary hyperparathyroidism is a cornerstone treatment for the prevention of bone fracture in patients with CKD24). Importantly, low bone turnover is also a risk factor for CVC. Decreased buffer function of low bone turnover often causes hypercalcemia and hyperphosphatemia, thereby accelerating CVC47). Accordingly, maintaining normal bone turnover is necessary for preventing CVC and cardiovascular death.
In this section, we will describe the contribution of each CKD–MBD mediator to the pathogenesis of CVD in patients with CKD. The influences of CKD–MBD mediators on the cardiovascular system are simplified in Fig.5.

In patients with CKD, as kidney function declines, relative phosphate overload develops and circulating concentrations of PTH and FGF23 increase, leading to the formation of CPPs. The serum concentrations of α-klotho, calcitriol, and calcidiol decrease. These abnormalities in CKD–MBD ultimately accelerate atherosclerosis, arteriosclerosis, hypertension, left ventricular hypertrophy, congestive heart failure, arrhythmia, and myocardial and cardiovascular calcification, all of which increase the risk of death.
CKD, chronic kidney disease; CKD–MBD, chronic kidney disease–mineral and bone disorder; CPPs, calciprotein particles; FGF23, fibroblast growth factor 23; PTG, parathyroid gland; PTH, parathyroid hormone.
Mounting evidence suggests that hyperphosphatemia is associated with an increased risk of cardiovascular events, including myocardial infarction, congestive heart failure, sudden death, brain hemorrhage, and peripheral arterial disease48-50). Hyperphosphatemia induces endothelial dysfunction by blunting nitric oxidase synthesis, which accelerates hypertension and atherosclerosis51). Hyperphosphatemia also drives CVC via the formation and maturation of CPPs and other cell-mediated calcification processes by sodium-phosphate co-transporters31, 35, 52). The association between hyperphosphatemia and cardiovascular events in clinical studies is partly mediated by increased PTH and FGF23 concentrations as a reaction to relative phosphate overload.
2) CalciumObservational studies have shown that hypercalcemia is a strong predictor of cardiovascular, infection-related, and all-cause mortality among hemodialysis patients23, 53-55).
Hypercalcemia accelerates CPP formation and maturation, eventually leading to the progression of CVC and atherosclerosis/arteriosclerosis31, 35, 56). Ectopic calcification in the cardiac conduction system induced by hypercalcemia increases the risk of bradyarrhythmia-related sudden death57, 58). Hypocalcemia is also associated with an increased risk of death, probably as a result of hypotension, congestive heart failure, and arrhythmia-related sudden death2, 48, 59). Hypocalcemia also causes QT-interval elongation, which increases the risk of ventricular tachycardia and ventricular fibrillation60, 61).
3) PTHA high serum PTH concentration is associated with an increased risk of cardiovascular death and bone fracture in dialysis-independent and dialysis-dependent CKD patients23, 55, 62, 63). PTH directly induces left ventricular hypertrophy by activating cardiomyocytes64). Surgical parathyroidectomy reduces the incidence of cardiovascular death65). PTH acts on PTH receptors on osteoblasts, thereby activating osteoclasts in bone and increasing calcium and phosphate release into the circulation, ultimately increasing the risk of hypercalcemia, hyperphosphatemia, and CVC66). In pre-dialysis CKD patients, the prevalence of atrial fibrillation may also be increased in subjects with high circulating PTH concentrations67).
4) FGF23Epidemiological studies have shown that a higher serum FGF23 concentration is associated with cardiovascular events and mortality in pre-dialysis CKD, kidney transplant, and hemodialysis patients68-70). Basic studies have revealed that FGF23 directly acts on cardiomyocytes and induces hypertrophy via the nuclear factor-kappa beta-calcineurin pathway71, 72). FGF23 also upregulates sodium–chloride co-transporters in the renal distal tubules and causes sodium chloride retention, thereby elevating the risk of volume overload and congestive heart failure73). Clinical studies have shown that increased circulating concentrations of FGF23 are associated with an increased risk of atrial fibrillation in patients with pre-dialysis CKD74). However, because FGF23 is not solely regulated by CKD–MBD but other factors, including iron deficiency, aldosterone, inflammation, cardiac damage, and ischemia, it is still controversial whether FGF23 per se has a direct impact on clinically relevant outcomes or whether confounding factors may account for the association between high serum FGF23 concentrations and worse clinical outcomes among CKD populations75).
5) α-KlothoMultiple cardioprotective effects have been reported with respect to α-klotho76-80). α-Klotho-deficient mice show cardiac dysfunction, cardiac fibrosis, and left ventricular hypertrophy, as well as overexpression of α-klotho-attenuated left ventricular hypertrophy in mice77, 78). α-Klotho inhibits nitric oxidase synthesis; therefore, α-klotho deficiency may cause hypertension79). The α-klotho concentration is also associated with the risk of atrial fibrillation80). A lower serum concentration of α-klotho is associated with an increased risk of cardiovascular events in patients with CKD81).
6) Calcitriol (1,25-Dihydroxyvitamin D)A literature review indicates that calcitriol has cardioprotective effects82, 83). Calcitriol inhibits renin expression, which leads to activation of the renin–angiotensin–aldosterone system, thereby causing cardiovascular damage82, 83). Vitamin D deficiency is also related to endothelial dysfunction, increased vascular stiffness, hypertension, left ventricular hypertrophy, and congestive heart failure82-84).
7) MagnesiumIonized magnesium is often decreased in patients with CKD85). Magnesium deficiency is related to CVD, including hypertension and CVC85). Magnesium has the potential to prevent the formation and maturation of CPPs, thereby possessing the capacity to halt CVC progression86, 87).
Two major pharmacological interventions for CKD–MBD in pre-dialysis CKD include phosphate binders and vitamin D receptor activators (VDRAs). An increasing number of randomized controlled trails (RCTs) are being conducted to examine the beneficial effects of CKD–MBD treatment. However, conflicting results have been reported in pre-dialysis CKD. As for phosphate binders, calcium carbonate, lanthanum carbonate, bixalomer, and ferric citrate are available to patients with pre-dialysis CKD in Japan. Phosphate binders reduce the serum phosphate and FGF23 concentrations88-90). However, vascular function is not improved by phosphate binders88, 90). As for CVC, one RCT showed that phosphate binders accelerate the progression of vascular calcification in individuals with pre-dialysis CKD91). By contrast, another RCT showed that sevelamer carbonate improved survival compared with calcium carbonate in patients with pre-dialysis CKD92). A recent pilot RCT showed that iron-based phosphate binders decrease the incidence of hospitalization and the composite outcome in patients with CKD stage G5 not undergoing dialysis93). The reasons for the improved outcome in patients treated with ferric citrate may be two-fold. First, iron supplementation with ferric citrate improves iron status, leading to an improvement in renal anemia, followed by a reduction in cardiovascular risk94). Another reason may be that reduced phosphate loading by ferric citrate reduces the phosphate-induced cardiovascular burden95). The beneficial effects of phosphate binders in pre-dialysis CKD are still debated.
Calcimimetics are not indicated in patients with pre-dialysis CKD; therefore, VDRAs are the unique treatment for uremic secondary hyperparathyroidism in this population96, 97). VDRAs reduce the serum PTH concentration by directly suppressing PTH synthesis and secretion, as well as indirectly via calcium loading mediated by increased intestinal calcium absorption. Pleiotropic effects of VDRAs, including the cardioprotective and reno-protective effects, have been reported98). However, VDRAs expose patients to an elevated risk of hypercalcemia, hypercalciuria, acute kidney injury, and CVC99). In fact, two RCTs—the PRIMO Trial and the OPERA Trial—failed to show the beneficial effects of VDRAs on cardiovascular function. VDRAs induced hypercalcemia more frequently than placebo in pre-dialysis CKD patients100, 101), although the subgroup analysis showed that patients treated with VDRAs developed congestive heart failure less frequently than those treated with placebo. Another RCT of vitamin D (cholecalciferol) supplementation demonstrated the protective effects on vascular function determined by flow-mediated dilatation in pre-dialysis CKD102). However, the beneficial effects of cholecalciferol and other VDRAs have not been proven in other RCTs103-105). At present, it may be too early to conclude that vitamin D or VDRAs should be administered to patients with pre-dialysis CKD. Intriguingly, 25-hydroxyvitamin D has been shown to suppress the serum PTH concentration and induce hypercalcemia less frequently than calcitriol, suggesting that 25-hydroxyvitamin D could be a therapeutic option for uremic secondary hyperparathyroidism in pre-dialysis CKD patients106).
Given the existing evidence, whether intervention for CKD–MBD in the pre-dialysis stage is beneficial or harmful remains controversial. Hence, further well-designed studies, including large-scale RCTs, are necessary to better understand the pathophysiology of CKD–MBD and identify appropriate treatment strategies for this condition.
2) Dialysis-Dependent CKDCompared with patients with pre-dialysis CKD, pharmacological approaches are more common in the dialysis population. Given that phosphate overload is the upstream pathology of CKD–MBD, phosphate unloading is reasonable. Phosphate binders are prescribed to patients with CKD to reduce the phosphate burden and lower the serum phosphate concentration by inhibiting phosphate absorption via the gastrointestinal tract107). Very recently, the EPISODE Trial, an RCT targeting maintenance hemodialysis patients with hyperphosphatemia, showed that strict phosphate control using phosphate binders reduced the progression of coronary arterial calcification compared with non-strict phosphate control, demonstrating that phosphate control using phosphate binders is effective to slow CVC progression108). At present, six types of phosphate binder are available on the Japanese market: calcium carbonate, sevelamer hydrochloride, bixalomer, lanthanum carbonate, ferric citrate, and sucroferric oxyhydroxide. Each phosphate binder has both advantages and disadvantages107, 109). Hence, the prescription of phosphate binders should be tailored to each patient depending on the patient’s background107). In recent years, calcium-based phosphate binders have been less commonly prescribed. This is because calcium loading is reported to accelerate the progression of CVC in the CKD population110). The KDIGO guidelines suggest that the dose of calcium-based phosphate binders should be reduced6, 7). Indeed, a network meta-analysis showed that sevelamer reduces the risk of death in patients undergoing hemodialysis compared with calcium-based phosphate binders111). However, the LANDMARK Trial, an RCT involving 2,374 Japanese hemodialysis patients with hyperphosphatemia, recently showed no significant survival benefit with lanthanum carbonate, another non-calcium-containing phosphate binder, compared with calcium carbonate112). Japanese hemodialysis patients consume a lower calcium-containing diet than those living in other countries; therefore, calcium-based phosphate binders might not increase the risk of CVD progression in these patients. However, whether non-calcium-based phosphate binders are better than calcium-based phosphate binders remains uncertain.
VDRAs are often used for the treatment of secondary hyperparathyroidism, vitamin D deficiency, and osteoporosis in patients undergoing dialysis113). VDRAs act on the parathyroid glands and suppress PTH synthesis and secretion, thereby decreasing the serum PTH concentration and reducing parathyroid gland volume19, 45). VDRAs also increase trans-intestinal calcium and phosphate absorption, elevate the serum concentrations of calcium and phosphate, and promote bone mineralization. For patients with hypocalcemia and who require positive calcium balance, VDRAs may be effective therapeutic options. In turn, excessive doses of VDRAs often cause hypercalcemia and hyperphosphatemia, potentially leading to CVC. A large-scale RCT conducted in Japan showed that low-dose VDRAs did not significantly reduce the incidence of cardiovascular events in Japanese patients with mild-to-moderate uremic secondary hyperparathyroidism among individuals undergoing maintenance hemodialysis114). Whether the beneficial effects of VDRAs on the health of hemodialysis patients are myth or fact remains uncertain.
Calcimimetics are allosteric modulators of calcium-sensing receptors that increase the sensitivity of these receptors to extracellular calcium, thereby inhibiting PTH secretion and synthesis. The reduction in the serum PTH concentration by calcimimetics lowers the serum calcium and phosphate concentrations115). The ADVANCE Trial and the EVOLVE Trial showed that calcimimetics have the potential to halt CVC progression and reduce the risk of all-cause mortality in patients undergoing hemodialysis116, 117). A sub-analysis of the EVOLVE Trial showed that calcimimetics lower the incidence of calciphylaxis in hemodialysis patients118). However, calcimimetics do have potential disadvantages, such as hypocalcemia. Hypocalcemia occasionally leads to lethal arrhythmia, hypotension, and heart failure. Therefore, hypocalcemia in patients treated with calcimimetics should be avoided119). Accordingly, the combination of calcimimetics with low-dose VDRAs is presumed to be a safe approach for the control of uremic secondary hyperparathyroidism. VDRAs increase the serum calcium concentration by increasing intestinal calcium absorption and suppressing PTH secretion120).
Patients with advanced uremic secondary hyperparathyroidism are potential candidates for surgical parathyroidectomy as a definitive treatment option19, 45). An observational study showed that surgical parathyroidectomy improved mortality in patients undergoing hemodialysis65). However, whether surgical parathyroidectomy is more preferable than calcimimetics remains uncertain121). The decision regarding which treatment option is appropriate for the control of uremic secondary hyperparathyroidism may be based on patients’ preferences and values, medical costs, and any undetermined clinical impact of both treatment options.
The dialysate calcium concentration is an under-recognized treatment option in patients undergoing dialysis. Higher calcium dialysate induces calcium overload and accelerates ectopic calcification, including CVC. Lower calcium dialysate can cause sudden cardiac death, especially when patients are treated with a lower potassium and bicarbonate dialysate. At present, dialysate calcium concentrations between 2.5 and 3.0 mEq/L are recommended for hemodialysis patients5-7). However, the optimal dialysate calcium concentration for maintenance hemodialysis patients should be explored in future studies122, 123).
CKD–MBD has emerged as an important complication in patients with CKD. Because CKD–MBD is a potent risk factor for cardiovascular morbidity and mortality, appropriate management of CKD–MBD is necessary to reduce the incidence of cardiovascular events and attain longevity in this population. At present, therapeutic options for the prevention of CKD–MBD are very limited in pre-dialysis CKD, whereas calcimimetics, phosphate binders, and VDRAs are cornerstones of treatment in patients undergoing dialysis. However, evidence on whether the treatment of CKD–MBD actually improves cardiovascular events and related death in patients with CKD is still insufficient. Therefore, further studies that provide a deeper understanding of the pathophysiology of CKD–MBD and its impact on the cardiovascular system are warranted for the prevention of CVD and to achieve a good prognosis among CKD populations.
We thank Emily Woodhouse, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
None.
SY and TN received lecture fees from the following pharmaceutical companies: Kyowa Kirin Co. Ltd., Kissei Pharmaceutical Co. Ltd., and Sanwa Kagaku Kenkyusho Co. Ltd.