Abstract
Metabolism is one of the vital functions of cells and living organisms, and the systems to sense and respond to the metabolic alterations play pivotal roles in a plethora of biological processes, including cell proliferative activities, immune cell functions, aging processes, and neuronal functions. Recently, we have reported that a transcriptional cofactor, C-terminal binding protein 2 (CtBP2), serves as a critical metabolite sensor in this context. CtBP2 has a structural pocket called Rossmann fold to accommodate metabolites, and it has been reported to be activated upon binding to NADH/NAD+. Owing to its preferential binding affinity for NADH compared with NAD+, increased glycolysis activates CtBP2 by regenerating NADH from NAD+. Furthermore, we recently reported that fatty acyl-CoAs, metabolites accumulated under the condition of lipid overload, as represented by obesity, can inactivate CtBP2. These observations suggest that CtBP2 monitors not only redox state but also energy substrate preference in the maintenance of metabolic homeostasis. In line with these metabolite-sensing capabilities, CtBP2 is activated in healthy subjects to protect against metabolic disturbances, whereas inactivation of CtBP2 in obesity contributes to the pathogeneses of obesity.
This metabolic system orchestrated by CtBP2 can provide a novel framework for understanding how cells maintain their homeostasis through coordination of metabolism, and CtBP2 incapacitation can be a critical point of the obesogenic cascade.
Intricate Metabolic Networks in Obesity Have to be Comprehensively Elucidated for the Formulation of Genuinely Effective Therapeutic Strategies
While obesity induces a wide spectrum of pathological conditions, the molecular networks behind this are intertwined and remain to be unraveled1-3). Obesity exerts a pervasive effect on all tissues and cells, with the liver serving as a prime example of metabolic complexity4). The unsuppressed gluconeogenesis in tandem with increased lipogenesis in the liver is one of the metabolic hallmarks of obesity5, 6). One of the widely used approaches to comprehend complex hepatic metabolic systems has been from hormonal perspectives as represented by the action of insulin. However, since potentiation of the action of insulin can suppress gluconeogenesis at the expense of lipogenesis, there has been a paradox and debate to understand obesity-induced metabolic abnormalities from this perspective5, 6). This gap in our knowledge of the mechanisms also limits the efficacy of our therapeutic approaches. Most of our current strategies to treat diabetes rely on the potentiation of the action of insulin that indeed ameliorates diabetes but aggravates hepatic steatosis. Since insulin also inhibits adipose tissue lipid mobilization, this therapeutic strategy also promotes obesity, leading to therapeutic impasse7).
To overcome this impasse, an alternative approach could be developed by targeting metabolites. Historically, substrates and end products have attracted attention in the understanding of the metabolic pathways. However, several metabolic intermediates generated throughout complex metabolic networks have emerged as multifunctional regulators that maintain cellular homeostasis, including epigenetic regulation, posttranslational modifications, and enzymatic activities8-11). One of the extensively investigated metabolites is NAD+ in part owing to its pro-longevity effect12). Sirtuins (SIRTs) are deacetylases whose catalytic activities, hydrolysis of acetyl-lysine moieties, are coupled with NAD+ breakdown into nicotinamide. In mammals, the sirtuin family comprises seven members (SIRT1-7), among which SIRT1 plays a central role in lifespan extension13). Since NAD+-dependent SIRT1 activation leads not only to lifespan extension but also to protection against a wide variety of diseases12-14), albeit with some exceptions15-17), this NAD+-driven system has been attracting attention from points of fundamental scientific inquiry as well as clinical exploitation. Alpha-ketoglutarate can be another example of multifunctional metabolites. It is a cofactor for some dioxygenase enzymes, including JmjC domain-containing histone demethylases (JHDMs) and ten–eleven translocation (TETs) demethylases18). Since alpha-ketoglutarate is generated by the enzymatic reaction of isocitrate dehydrogenases (IDHs) and glutamate dehydrogenase, epigenetic regulations controlled by JHDMs and TETs are under the influences of these metabolic activities19). Furthermore, mutations in IDHs frequently observed in some populations of cancer cells lead to the generation of 2-hydroxyglutarate instead of alpha-ketoglutarate, one of the metabolic hallmarks of cancer cells20, 21). Along with the growing interest in these metabolite-driven systems, a recent report comprehensively screened the metabolite–protein interactions and observed the unexpected regulation of lactate dehydrogenase by fatty acyl-CoAs22).
To mitigate the prevalence of obesity and the associated comorbidities from this metabolite-dependent perspective, we have investigated the roles of C-terminal binding protein 2 (CtBP2), a transcriptional cofactor harboring metabolite-sensing capabilities. There exist two isoforms of CtBPs in mammals, CtBP1 and CtBP2, with both having structural pocket called Rossmann fold to accommodate metabolites23, 24). The two major differences between these isoforms are subcellular localization and phenotypes observed in the global genetic deficiency. Only CtBP2 has an N-terminal nuclear localization signal and is exclusively localized to the nucleus, whereas CtBP1 distributes throughout the cells, indicating that the former is more specialized for transcription25). The global deficiency of CtBP2 manifests embryonic lethality, whereas that of CtBP1 exhibits milder phenotype26). The deficiency of both isoforms causes developmental defects, which in part explains the reasons that the roles of CtBPs have been underexplored. Since CtBPs are transcriptional cofactors lacking DNA-binding capability, CtBPs gain access to genomic regions through binding to their target transcription factors where they recruit epigenetic modifiers to alter chromatin architecture23, 24). The interaction between CtBPs and transcription factors is largely dependent on the multimerization of CtBPs. Upon binding to NADH/NAD+, CtBPs adopt dimer or multimer configurations27), which promote the transcriptional complex formation with some exceptions28, 29). It has been reported that the affinity of CtBPs for NADH is 100-fold higher than that for NAD+, indicating that CtBP2 can sense cellular redox state. In terms of their localization, CtBPs sense the NADH/NAD+ ratio in nuclear–cytosolic compartments which reflects the enzymatic activities of key metabolic system such as glycolysis and lactate dehydrogenase activity30). Despite these profound involvements in metabolic systems, investigations focusing on the metabolism or in vivo function of CtBPs have not been previously described.
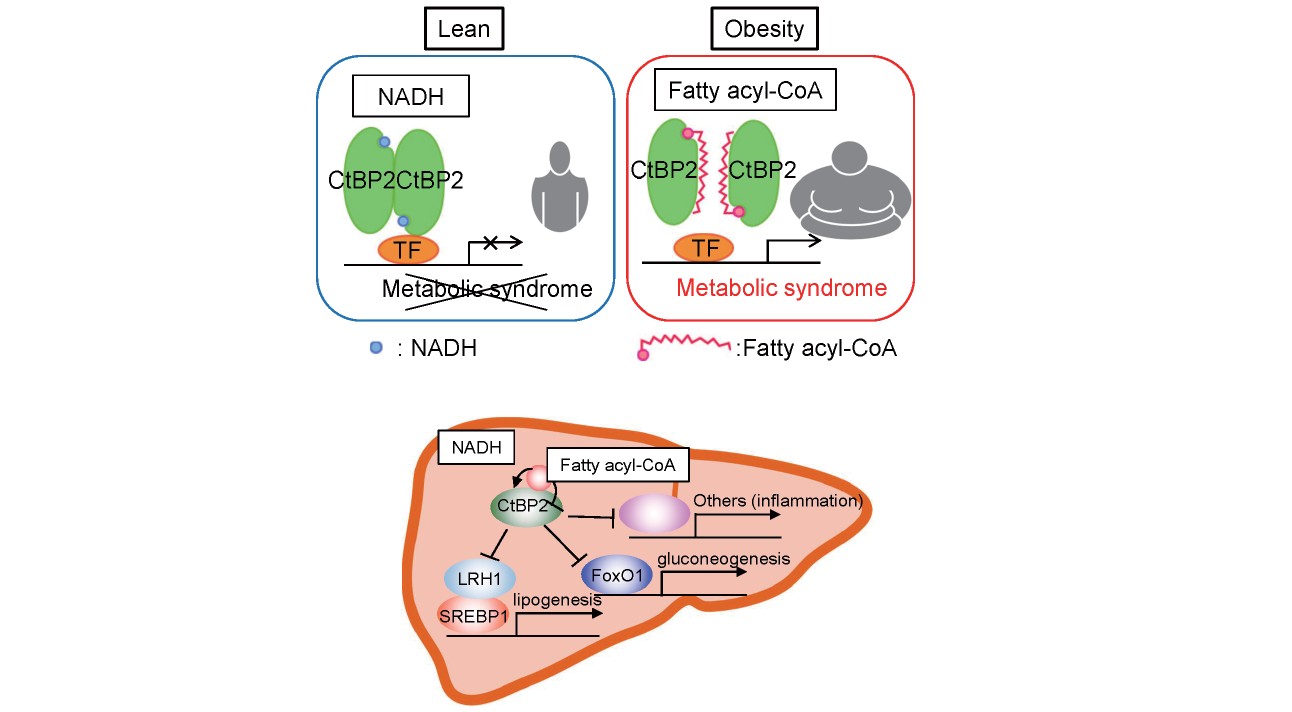
CtBP2 Plays Pivotal Roles in the Pathogenesis of Obesity
As aforementioned, the concurrent increase in gluconeogenesis and lipogenesis in the liver represents the complex nature of obesity-induced metabolic abnormalities. Recently, we demonstrated that CtBP2 inactivation in obesity can be one of the missing pieces of this enigmatic hepatic pathology31). We found that CtBP2 can be inactivated upon binding to fatty acyl-CoAs that are increased in the tissues in obese subjects. From a structural perspective, the CoA moiety of fatty acyl-CoAs, which shares adenosine structure with NADH/NAD+, competes the Rossmann fold pocket with NADH/NAD+, whereas the acyl-chain moiety resides at the interface of CtBP2 dimer to physically block CtBP2 dimerization. Together with the preceding observations that CtBP2 can be activated upon binding to NADH/NAD+, the dual specificity establishes CtBP2 as a metabolite sensor that responds to obesity-induced metabolic milieu (Fig.1). In addition, we found that CtBP2 directly interacts with Forkhead box O1 (FoxO1), a master transcription factor for hepatic gluconeogenesis32), to repress its function. Hepatic lipogenesis driven by sterol regulatory element-binding protein 1 (SREBP1)33) is also repressed by CtBP2 through a CtBP2/liver X receptor (LXR)/SREBP1 complex, as was further ensured by another independent report34). Intriguingly, these CtBP2/FoxO1 and CtBP2/SREBP1 complex formations were significantly diminished in mouse models of obesity and in human specimens, indicating that liberation of FoxO1 and SREBP1 from CtBP2-mediated repression is one of the key mechanisms underlying the concurrent activation of hepatic gluconeogenesis and lipogenesis in obesity (Fig.1). Contrarily, CtBP2 overexpression ameliorates diabetes and hepatic steatosis in obese mice, indicating the therapeutic potentials of targeting CtBP2. In line with our model, the beneficial effects of CtBP2 overexpression can be abolished by mutations in the metabolite-sensing pocket Rossmann fold and increased by a mutation at the dimeric interface that promotes CtBP2 dimer formation31).
We further extended our research to explore the possible involvement of peroxisome proliferator activator receptor alpha (PPARα) in this system, a master transcription factor for fatty acid oxidation29, 35). We found that CtBP2 indirectly binds to PPARα to repress the activity. Interestingly, in sharp contrast to the CtBP2/FoxO1 and CtBP2/SREBP1 complexes, this repressor complex formation is promoted in obesity, contributing to the obesity-induced reduction of fatty acid oxidation. In most cases, CtBP2 dimer/multimer preferentially binds to its target transcription factors, but in this case, it is the CtBP2 monomer that binds to PPARα. This study shed light on another aspect of the regulatory mechanisms of fatty acid oxidation. One of the acyl-CoA derivatives, malonyl-CoA, has been known to inhibit carnitine palmitoyltransferase 1 (CPT1) activity36), contributing to the negative regulation of fatty acid oxidation by preventing entry of fatty acid into the mitochondria. Since acyl-CoA binding shifts the monomer–dimer equilibrium of CtBP2 to monomer, malonyl-CoA accumulation leads to CtBP2 monomerization and PPARα repression, another layer of regulatory mechanisms of fatty acid oxidation29).
The widespread expression of CtBP2 across diverse tissues prompted us to investigate the potential roles of CtBP2 in nonhepatic tissues. Next, we examined pancreatic β-cells and found that CtBP2 is required to maintain normal β-cell function (Fig.2)37). In β-cells, CtBP2 forms a large transcriptional complex with Neuronal differentiation 1 (NEUROD1), a master transcription factor for the maintenance of β-cell integrity38, 39). The large CtBP2 transcriptional complex contains chromatin remodelers such as KMT2 (lysine methyltransferase 2) to maintain an open chromatin conformation in the promoters of key genes safeguarding β-cell functions. Interestingly, upon exposure to oxidative stress, the CtBP2 protein undergoes modification with polyubiquitin chains that destines proteins for degradation. Since pancreatic β-cells are extremely vulnerable to oxidative insults owing to their insufficient antioxidative defense system, and obesity provokes oxidative stress in a wide range of tissues, CtBP2 protein expression is significantly diminished in pancreatic β-cells in obesity. These findings led us to develop pancreatic β-cell-specific CtBP2 knockout models that manifest glucose intolerance due to impaired insulin secretion. These data support our model where the progressive decline of pancreatic β-cell function in obesity is induced at least in part by CtBP2 degradation37). It is noteworthy that loss of the function of CtBP2 in obese liver is caused by allosteric conformational change of CtBP2 upon binding to fatty acyl-CoA, whereas that in pancreatic β-cells in obesity is caused by the protein degradation of CtBP2. It is of additional interest that CtBP2 protein is quite vulnerable to oxidative insults, whereas the other isoform CtBP1 is insensitive to those, suggesting that CtBP2 is more preferentially involved in the pathogenesis of obesity37).

As aforementioned, CtBP2 serves as a sensor for redox state, and CtBP2 protein is vulnerable to oxidative stress, indicating that CtBP2 is a core component in the redox systems. Consistent with this idea, we also found that CtBP2 potentiates the antioxidative defense system by forming complexes with nuclear factor erythroid 2-related factor 1/2 (NFE2L1 and NFE2L2, also known as NRF1 and NRF2)40). The widely accepted view of the regulatory mechanisms behind NRF1 and NRF2 activities has focused on nuclear-cytosolic shuttling system. NRF1, embedded in the ER membrane, has been postulated to be regulated by intermembrane proteolysis that allows nuclear localization of NRF1 41, 42). NRF2 is sequestered in the cytoplasm by Kelch-like ECH-associated protein 1 (KEAP1) that facilitates polyubiquitination-dependent proteasomal degradation under normal conditions, whereas the sulfhydryl groups of cysteine residues in KEAP1 are oxidized upon oxidative insults, resulting in the liberation of NRF2 for nuclear translocation43). This study highlights that CtBP2 confers additional layer of regulation on these transcription factors in the nucleus. These findings suggest that CtBP2 can manage oxidative stress through this mechanism or others to some extent, although this system can be overwhelmed by massive oxidative damage that degrades CtBP2 protein.
Obesity-associated metabolic abnormalities can be classified into two categories: tissue-specific ones and systemic aberrations pervasive across tissues. While the former is currently under our investigation, the latter encompasses oxidative stress and inflammation. Intriguingly, the anti-inflammatory effects of CtBP2 activation have been repeatedly reported31, 44, 45). Chronic low-grade inflammation, alternatively called metaflammation2), is one of the hallmarks of obesity. Proper resolution of inflammation has been attracting broad interests, such as osteoarthritis, inflammatory bowel diseases, and even aging-related diseases. Senescent cells secrete proinflammatory cytokines that significantly contribute to the pathogenesis of age-related diseases46).
We also reported an involvement of CtBP2 in the endothelial mesenchymal transition (EMT) processes through direct interactions with the Pit-Oct-Unc (POU) domain transcription factors, OCT1 and OCT2 47). Although activation of the EMT program has been extensively investigated in the processes of tumor cell invasion and metastasis48), this program is inherently equipped in normal cells and controls a wide range of adaptive cellular processes, such as wound healing, fibrosis, and aging49). In pancreatic endocrine cells, the EMT has been reported to regulate transdifferentiation of α cells to β cells50). Therefore, CtBP2 may maintain proper β cell functions by also activating EMT.
Not only our reports but also others demonstrated the essential role of CtBP2 in other tissues and cells: Meijer et al. reported that CtBP2 is required to maintain intestinal stem cells against ER stress51). It was also reported that CtBP2 in neuronal stem and progenitor cells plays a crucial role in the cortical development in mouse brain52).
Historical Perspective and Future Directions
Historically, the roles of CtBPs have been investigated mainly in the context of cell proliferative activities and cancer progression23, 24). Majority of the studies support the tumor-promoting roles of CtBPs, albeit with exceptions53, 54). However, there may be some potential pitfalls in this issue. Many studies did not discriminate between CtBP1 and CtBP2 in their analyses. We summarize the key differences between CtBP1 and CtBP2 in Table 1 that we need to be aware of. Furthermore, most of the preceding studies drew their conclusion based on the effects of the overexpression and/or knockdown/genetic deletion of CtBPs that do not faithfully recapitulate allosteric alterations of CtBP activities. Furthermore, the antioxidative properties of CtBP2 would confer survival advantages to cancer cells but may not initiate tumorigenesis. We and others reported the involvement of CtBP2 in the EMT program47, 55) that plays a critical role in cancer cell biology. However, cells without any malignant potential do not use this program for uncontrolled migration and invasion; rather they activate it for wound healing or tissue repair49). In parallel, this perspective may play an important role in the lineage commitment of endocrine cells in pancreas50). Indeed, our transcriptomic analysis of CtBP2 overexpression in the liver did not reveal any signs of increased cell proliferative activities. Since CtBP2 activation would confer protection against obesity and its associated diseases, this issue should be thoroughly addressed in future investigations. In addition, while we reported the inactivation of CtBP2 in obesity and the metabolic benefits achieved by CtBP2 activation in the liver and pancreatic β-cells, the potential roles of CtBP2 in other tissues and cells await future research. Despite these uncertainties, it is a plausible and attractive possibility to identify CtBP2-activating small molecule(s) targeting the metabolite-sensing pocket Rossmann fold. In pancreatic β-cells, CtBP2 inactivation is regulated at the protein expression levels. However, since the CtBP2 protein can be stabilized by accommodation of NADH/NAD+, pharmacological targeting of the Rossmann fold could also protect the CtBP2 protein against degradation31).
Table 1.Major differences hitherto identified between CtBP1 and CtBP2
|
CtBP1 |
CtBP2 |
| Rossmann fold pocket23, 24)
|
Equipped |
Equipped |
| Intracellular localization23, 24)
|
Throughout the cells (mainly in the cytosol and nucleus) |
Mainly in nucleus |
| Global knockout phenotype26)
|
Viable with developmental defects |
Embryonic lethal |
| Oxidative stress induced protein degradation |
Insensitive |
Sensitive |
CtBP2 has been recognized as a redox sensor based on the capability to respond to the NADH/NAD+ ratio56). In addition to this pyridine dinucleotide-sensing capability, we found that CtBP2 can accommodate fatty acyl-CoAs to adopt the monomer configuration in contrast to the dimer formation with NADH/NAD+31). This dual specificity can be explained from a structural point of view: NADH/NAD+ and the CoA moiety of fatty acyl-CoAs share adenosine structure that can be accommodated in the Rossmann fold pocket of CtBP2. CtBP2 would bind to other adenosine derivatives, while there seems to be some specificities for adenosine derivatives based on the fact that NADPH/NADP+, harboring phosphate group, lacks binding affinity for CtBP2 56). As regards fatty acyl-CoAs, fatty acyl-CoAs with longer acyl chain exhibit higher affinities for CtBP2 and more potently inhibit activities of CtBP2 in line with our structural modeling29, 31). There may exist potentially important adenosine derivatives that are unidentified with biological significance.
In part because the metabolites capable of modulating the activity of CtBP2 remain to be fully cataloged, the roles of CtBP2 in physiology and pathology are still underexplored. The acyl-CoA binding properties confer a new framework for the understanding of metabolism with a focused attention to CtBP2. Fatty acyl-CoA accumulation may be caused by impaired lipid dissipation, increased de novo lipogenesis, or increased lipid influx. While CtBP2 may discriminate fatty acyl-CoAs from different sources, this fatty acyl-CoA-sensing capability of CtBP2 indicates that CtBP2 monitors cellular fatty acid metabolism. On the other hand, the pyridine dinucleotide-sensing capability of CtBP2 also indicates that CtBP2 monitors glycolytic activities. Living organisms rely on energy substrate utilization to fulfill their energy requirements and balance between anaerobic glycolysis and aerobic oxidative phosphorylation depending on the metabolic milieus comprised of oxygen, energy substrates, and other constituents. CtBP2 would be a sensor that monitors the coordination of cellular energy metabolism. The balance between glycolysis and oxidative phosphorylation is a key determinant for numerous biological processes, such as immune cell polarization57) and cancer cell survival/transformation58). Thus, CtBP2 would play critical roles in a wide variety of biological processes, and there may remain unidentified functions that await future investigation.
Conclusion
We recently reported the intriguing involvement of CtBP2 in the pathogenesis of obesity, which may pave a way for therapeutic breakthroughs. These works together with preceding reports present a novel framework for understanding the complex biology in obesity and diverse biological systems. Future investigations should reveal unanticipated insights and captivating discoveries.
Acknowledgements
We thank the members of the Shimano laboratory for their contributions and invaluable discussions. M.S. was supported by Japan Promotion of Science (Grant Number 20K08855, 23K18270), the Japan Agency for Medical Research and Development (AMED) under Grant Number JP18gm5910007, JP22ek0210175, and JP22gm6710004, Takeda Science Foundation, Ono Medical Research Foundation and Japan Diabetes Foundation. MS received the Goto Yuichiro Award at the 55th Annual Scientific Meeting of the Japan Atherosclerosis Society, and this review paper was written to expound upon the significant contributions and novel insights presented during the conference.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1) Shulman GI: Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med, 2014; 371: 2237-2238
- 2) Hotamisligil GS: Inflammation, metaflammation and immunometabolic disorders. Nature, 2017; 542: 177-185
- 3) Rohm TV, Meier DT, Olefsky JM, Donath MY: Inflammation in obesity, diabetes, and related disorders. Immunity, 2022; 55: 31-55
- 4) Lim S, Kim JW, Targher G: Links between metabolic syndrome and metabolic dysfunction-associated fatty liver disease. Trends Endocrinol Metab, 2021; 32: 500-514
- 5) Brown MS, Goldstein JL: Selective versus total insulin resistance: a pathogenic paradox. Cell Metab, 2008; 7: 95-96
- 6) Sun Z, Lazar MA: Dissociating fatty liver and diabetes. Trends Endocrinol Metab, 2013; 24: 4-12
- 7) Heller S: Weight gain during insulin therapy in patients with type 2 diabetes mellitus. Diabetes Res Clin Pract, 2004; 65 Suppl 1: S23-27
- 8) Kinnaird A, Zhao S, Wellen KE, Michelakis ED: Metabolic control of epigenetics in cancer. Nat Rev Cancer, 2016; 16: 694-707
- 9) Wang ZA, Cole PA: The Chemical Biology of Reversible Lysine Post-translational Modifications. Cell Chem Biol, 2020; 27: 953-969
- 10) Diskin C, Ryan TAJ, O’Neill LAJ: Modification of Proteins by Metabolites in Immunity. Immunity, 2021; 54: 19-31
- 11) Martínez-Reyes I, Chandel NS: Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun, 2020; 11: 102
- 12) Bonkowski MS, Sinclair DA: Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol, 2016; 17: 679-690
- 13) Haigis MC, Guarente LP: Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev, 2006; 20: 2913-2921
- 14) Lavu S, Boss O, Elliott PJ, Lambert PD: Sirtuins--novel therapeutic targets to treat age-associated diseases. Nat Rev Drug Discov, 2008; 7: 841-853
- 15) Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P: Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature, 2005; 434: 113-118
- 16) Deng CX: SIRT1, is it a tumor promoter or tumor suppressor? Int J Biol Sci, 2009; 5: 147-152
- 17) Hu Y, Liu J, Wang J, Liu Q: The controversial links among calorie restriction, SIRT1, and resveratrol. Free Radic Biol Med, 2011; 51: 250-256
- 18) Baksh SC, Finley LWS: Metabolic Coordination of Cell Fate by α-Ketoglutarate-Dependent Dioxygenases. Trends Cell Biol, 2021; 31: 24-36
- 19) Abla H, Sollazzo M, Gasparre G, Iommarini L, Porcelli AM: The multifaceted contribution of α-ketoglutarate to tumor progression: An opportunity to exploit? Semin Cell Dev Biol, 2020; 98: 26-33
- 20) Prensner JR, Chinnaiyan AM: Metabolism unhinged: IDH mutations in cancer. Nat Med, 2011; 17: 291-293
- 21) Pirozzi CJ, Yan H: The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol, 2021; 18: 645-661
- 22) Hicks KG, Cluntun AA, Schubert HL, Hackett SR, Berg JA, Leonard PG, Ajalla Aleixo MA, Zhou Y, Bott AJ, Salvatore SR, Chang F, Blevins A, Barta P, Tilley S, Leifer A, Guzman A, Arok A, Fogarty S, Winter JM, Ahn HC, Allen KN, Block S, Cardoso IA, Ding J, Dreveny I, Gasper WC, Ho Q, Matsuura A, Palladino MJ, Prajapati S, Sun P, Tittmann K, Tolan DR, Unterlass J, VanDemark AP, Vander Heiden MG, Webb BA, Yun CH, Zhao P, Wang B, Schopfer FJ, Hill CP, Nonato MC, Muller FL, Cox JE, Rutter J: Protein-metabolite interactomics of carbohydrate metabolism reveal regulation of lactate dehydrogenase. Science, 2023; 379: 996-1003
- 23) Chinnadurai G: Transcriptional regulation by C-terminal binding proteins. Int J Biochem Cell Biol, 2007; 39: 1593-1607
- 24) Stankiewicz TR, Gray JJ, Winter AN, Linseman DA: C-terminal binding proteins: central players in development and disease. Biomol Concepts, 2014; 5: 489-511
- 25) Bergman LM, Morris L, Darley M, Mirnezami AH, Gunatilake SC, Blaydes JP: Role of the unique N-terminal domain of CtBP2 in determining the subcellular localisation of CtBP family proteins. BMC Cell Biol, 2006; 7: 35
- 26) Hildebrand JD, Soriano P: Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol Cell Biol, 2002; 22: 5296-5307
- 27) Bellesis AG, Jecrois AM, Hayes JA, Schiffer CA, Royer WE, Jr.: Assembly of human C-terminal binding protein (CtBP) into tetramers. J Biol Chem, 2018; 293: 9101-9112
- 28) Bhambhani C, Chang JL, Akey DL, Cadigan KM: The oligomeric state of CtBP determines its role as a transcriptional co-activator and co-repressor of Wingless targets. Embo j, 2011; 30: 2031-2043
- 29) Saito K, Sekiya M, Kainoh K, Yoshino R, Hayashi A, Han SI, Araki M, Ohno H, Takeuchi Y, Tsuyuzaki T, Yamazaki D, Wanpei C, Hada L, Watanabe S, Paramita Adi Putri PI, Murayama Y, Sugano Y, Osaki Y, Iwasaki H, Yahagi N, Suzuki H, Miyamoto T, Matsuzaka T, Shimano H: Obesity-induced metabolic imbalance allosterically modulates CtBP2 to inhibit PPAR-alpha transcriptional activity. J Biol Chem, 2023; 299: 104890
- 30) Kim JW, Dang CV: Multifaceted roles of glycolytic enzymes. Trends Biochem Sci, 2005; 30: 142-150
- 31) Sekiya M, Kainoh K, Sugasawa T, Yoshino R, Hirokawa T, Tokiwa H, Nakano S, Nagatoishi S, Tsumoto K, Takeuchi Y, Miyamoto T, Matsuzaka T, Shimano H: The transcriptional corepressor CtBP2 serves as a metabolite sensor orchestrating hepatic glucose and lipid homeostasis. Nat Commun, 2021; 12: 6315
- 32) Accili D, Arden KC: FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell, 2004; 117: 421-426
- 33) Shimano H, Sato R: SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol, 2017; 13: 710-730
- 34) Ferrarese R, Izzo A, Andrieux G, Lagies S, Bartmuss JP, Masilamani AP, Wasilenko A, Osti D, Faletti S, Schulzki R, Yuan S, Kling E, Ribecco V, Heiland DH, Tholen S, Prinz M, Pelicci G, Kammerer B, Boerries M, Carro MS: ZBTB18 inhibits SREBP-dependent lipid synthesis by halting CTBPs and LSD1 activity in glioblastoma. Life Sci Alliance, 2023; 6
- 35) Wahli W, Michalik L: PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab, 2012; 23: 351-363
- 36) Foster DW: Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J Clin Invest, 2012; 122: 1958-1959
- 37) Sekiya M, Ma Y, Kainoh K, Saito K, Yamazaki D, Tsuyuzaki T, Chen W, Adi Putri PIP, Ohno H, Miyamoto T, Takeuchi Y, Murayama Y, Sugano Y, Osaki Y, Iwasaki H, Yahagi N, Suzuki H, Motomura K, Matsuzaka T, Murata K, Mizuno S, Takahashi S, Shimano H: Loss of CtBP2 may be a mechanistic link between metabolic derangements and progressive impairment of pancreatic β cell function. Cell Rep, 2023: 112914
- 38) Chakrabarti SK, Mirmira RG: Transcription factors direct the development and function of pancreatic beta cells. Trends Endocrinol Metab, 2003; 14: 78-84
- 39) van der Meulen T, Huising MO: Role of transcription factors in the transdifferentiation of pancreatic islet cells. J Mol Endocrinol, 2015; 54: R103-117
- 40) Kainoh K, Takano R, Sekiya M, Saito K, Sugasawa T, Ma Y, Murayama Y, Sugano Y, Osaki Y, Iwasaki H, Takeuchi Y, Yahagi N, Suzuki H, Miyamoto T, Nakagawa Y, Matsuzaka T, Shimano H: CtBP2 confers protection against oxidative stress through interactions with NRF1 and NRF2. Biochem Biophys Res Commun, 2021; 562: 146-153
- 41) Radhakrishnan SK, den Besten W, Deshaies RJ: p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. Elife, 2014; 3: e01856
- 42) Koizumi S, Irie T, Hirayama S, Sakurai Y, Yashiroda H, Naguro I, Ichijo H, Hamazaki J, Murata S: The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. Elife, 2016; 5
- 43) Baird L, Yamamoto M: The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol Cell Biol, 2020; 40
- 44) Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK: An ADIOL-ERβ-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell, 2011; 145: 584-595
- 45) Shen Y, Kapfhamer D, Minnella AM, Kim JE, Won SJ, Chen Y, Huang Y, Low LH, Massa SM, Swanson RA: Bioenergetic state regulates innate inflammatory responses through the transcriptional co-repressor CtBP. Nat Commun, 2017; 8: 624
- 46) Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F: Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol, 2021; 22: 75-95
- 47) Ma Y, Sekiya M, Kainoh K, Matsuda T, Iwasaki H, Osaki Y, Sugano Y, Suzuki H, Takeuchi Y, Miyamoto T, Yahagi N, Nakagawa Y, Matsuzaka T, Shimano H: Transcriptional co-repressor CtBP2 orchestrates epithelial-mesenchymal transition through a novel transcriptional holocomplex with OCT1. Biochem Biophys Res Commun, 2020; 523: 354-360
- 48) Shibue T, Weinberg RA: EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol, 2017; 14: 611-629
- 49) Lamouille S, Xu J, Derynck R: Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol, 2014; 15: 178-196
- 50) Ben-Othman N, Vieira A, Courtney M, Record F, Gjernes E, Avolio F, Hadzic B, Druelle N, Napolitano T, Navarro-Sanz S, Silvano S, Al-Hasani K, Pfeifer A, Lacas-Gervais S, Leuckx G, Marroquí L, Thévenet J, Madsen OD, Eizirik DL, Heimberg H, Kerr-Conte J, Pattou F, Mansouri A, Collombat P: Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-like Cell Neogenesis. Cell, 2017; 168: 73-85.e11
- 51) Meijer BJ, Smit WL, Koelink PJ, Westendorp BF, de Boer RJ, van der Meer JHM, Vermeulen JLM, Paton JC, Paton AW, Qin J, Dekker E, Muncan V, van den Brink GR, Heijmans J: Endoplasmic reticulum stress regulates the intestinal stem cell state through CtBP2. Sci Rep, 2021; 11: 9892
- 52) Karaca E, Li X, Lewicki J, Neofytou C, Guérout N, Barnabé-Heider F, Hermanson O: The corepressor CtBP2 is required for proper development of the mouse cerebral cortex. Mol Cell Neurosci, 2020; 104: 103481
- 53) Sierra J, Yoshida T, Joazeiro CA, Jones KA: The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev, 2006; 20: 586-600
- 54) Subramanian T, Zhao LJ, Chinnadurai G: Interaction of CtBP with adenovirus E1A suppresses immortalization of primary epithelial cells and enhances virus replication during productive infection. Virology, 2013; 443: 313-320
- 55) Chinnadurai G: The transcriptional corepressor CtBP: a foe of multiple tumor suppressors. Cancer Res, 2009; 69: 731-734
- 56) Zhang Q, Piston DW, Goodman RH: Regulation of corepressor function by nuclear NADH. Science, 2002; 295: 1895-1897
- 57) Jung J, Zeng H, Horng T: Metabolism as a guiding force for immunity. Nat Cell Biol, 2019; 21: 85-93
- 58) Faubert B, Solmonson A, DeBerardinis RJ: Metabolic reprogramming and cancer progression. Science, 2020; 368
