Abstract
Aim: This study aimed to analyze two cases of marked hypo-high-density lipoprotein (HDL) cholesterolemia to identify mutations in ATP-binding cassette transporter A1 (ABCA1) and elucidate the molecular mechanism by which these novel pathological mutations contribute to hypo-HDL cholesterolemia in Tangier disease.
Methods: Wild type and mutant expression plasmids containing a FLAG tag inserted at the C-terminus of the human ABCA1 gene were generated and transfected into HEK293T cells. ABCA1 protein expression and cholesterol efflux were evaluated via Western blotting and efflux assay. The difference in the rate of change in protein expression was evaluated when proteolytic and protein-producing systems were inhibited.
Results: In case 1, a 20-year-old woman presented with a chief complaint of gait disturbance. Her HDL-C level was only 6.2 mg/dL. Tangier disease was suspected because of muscle weakness, decreased nerve conduction velocity, and splenomegaly. Whole-exome analysis showed compound heterozygosity for a W484* nonsense mutation and S1343I missense mutation, which confirmed Tangier disease. Cholesterol efflux decreased by a mixture of W484* and S1343I mutations. The S1343I mutation decreased the protein production rate but increased the degradation rate, decreasing the protein levels. This patient also had Krabbe disease. The endogenous ABCA1 protein level of macrophage cell decreased by knocking down its internal galactocerebrosidase.
Case 2, a 51-year-old woman who underwent tonsillectomy presented with peripheral neuropathy, corneal opacity, and HDL-C of 3.4 mg/dL. Whole-exome analysis revealed compound heterozygosity for R579* and R1572* nonsense mutations, which confirmed Tangier disease.
Conclusion: Case 1 is a new ABCA1 mutation with complex pathogenicity, namely, a W484*/S1343I compound heterozygote with marked hypo-HDL cholesterolemia. Analyses of the compound heterozygous mutations indicated that decreases in ABCA1 protein levels and cholesterol efflux activity caused by the novel S1343I mutation combined with loss of W484* protein activity could lead to marked hypo-HDL cholesterolemia. Galactocerebrosidase dysfunction could also be a potential confounding factor for ABCA1 protein function.
Introduction
ATP-binding cassette transporter A1 (ABCA1) is a 12-transmembrane ATP-binding cassette protein on the plasma membrane and has two nucleotide-binding domains (NBD-1 and NBD-2). The two extracellular loops are disulfide-linked and are involved in ATP and apolipoprotein A1 (ApoA1) binding and cholesterol efflux1-4).
ApoA1, which is synthesized in the liver and small intestine, binds to free cholesterol (FC) by the action of ABCA1 in peripheral tissues to form nascent high-density lipoprotein (HDL). The nascent HDL then acts on ABCA1 on the cell surface of peripheral tissues to draw out intracellular FC, which is then transferred to the nascent HDL by lecithin cholesterol acyltransferase (LCAT). The FC accumulated on the surface of HDL particles is converted to cholesteryl ester (CE) by the action of LCAT and accumulates from the surface to the center of HDL to form mature HDL. Cholesteryl ester transfer protein (CETP) transfers the CE accumulated in HDL to low-density lipoprotein (LDL) (HDL catabolism)5-9).
Tangier disease is an autosomal recessive genetic disorder in which serum HDL-cholesterol levels are significantly low because of decreased cholesterol efflux. It is also associated with ABCA1 defects. CEs accumulate in the liver, spleen, peripheral nerves, and other organs. Tangier disease is rare worldwide. Serum HDL levels are reduced to zero in homozygous mutants and to normal or mildly below normal levels in heterozygous mutants. Typical symptoms of Tangier disease include enlarged tonsils with a characteristic orange appearance, hepatomegaly or splenomegaly, corneal opacity, and peripheral neuropathy. Tangier disease clinically causes juvenile coronary artery disease, which has a poor prognosis10-15).
Herein, we experienced two such cases in a 20-year-old and a 51-year-old female patient. They presented with marked hypo-HDL cholesterolemia and were finally diagnosed with Tangier disease.
Aim
This study aimed to evaluate the presence or absence of ABCA1 mutations by analyzing two patients with marked hypo-HDL cholesterolemia and elucidate the molecular mechanism by which these novel mutations contribute to hypo-HDL cholesterolemia as a pathological variant in Tangier disease.
Methods
1. Reagents
Recombinant human ApoA1 (REF 019-20731), MG132 (REF 139-18451), actinomycin D (REF 018-21264), and cycloheximide (REF 037-20991) were purchased from Fujifilm Wako Pure Chemicals Corporation (Japan). In addition, TopFluor® Cholesterol 23-(dipyrrometheneboron difluoride)-24-norcholesterol (REF 810255) was purchased from Avanti Polar Lipids (AL, USA), KOD-Plus-(KOD-201) from Toyobo (Japan), Opti-MEM® (11058-021) from Gibco (MA, USA), and FuGENE® HD Transfection Reagent (E231A) from Promega Corporation (WI, USA). Monoclonal Anti-FLAG M2 antibody produced in mouse (F3165-1MG) was purchased from Sigma-Aldrich (MO, USA), ABCA1 antibody (MA5-16026) from Invitrogen (MA, USA), Anti-GALC antibody (11991-1-AP) from Proteintech Group (IL, USA), and Anti-mouse IgG, HRP-linked antibody (7076S) from Cell Signaling Technology (MA, USA). Calpeptin (REF S7396) was purchased from Selleck Chemicals (TX, USA), Sepasol-RNA I Super G (09379-55) from Nacalai Tesque, Inc. (Japan), and PrimeScriptTM RT Master Mix (RR036A) and TB Green® Premix Ex TaqTM II (RR820A) from Takara (Japan). Dulbecco’s Modified Eagle Medium (DMEM) was purchased from Gibco (31053-028), and RPMI 1640 with glutamine liquid (30264-56), 12-O-Tetradecanoyl Phorbol 13-Acetate (PMA) (27547-14), and bovine serum albumin (general grade, pH 7.0) (01860-65) were purchased from Nacalai Tesque, Inc. The plasmid human ABCA1-FLAG in pcDNA3.1 was prepared by Dr. Hirano16).
2. Preparation of the Homemade Constructs
Nonsense mutant plasmids (W484*, R579*, and R1572*) were produced as follows:
W484* and R579*: pcDNA 3.1(+) was cut with restriction enzymes AflII and ApaI, and the AflII to ApaI polymerase chain reaction (PCR) product was inserted with a FLAG sequence (GACTACAAGGACGATGACGACAAG) just before the stop mutation of hABCA1. The primer sequence of the inserted sequence was as follows:
W484*
Forward: AGCGTTTAAACTTAAGATGGCTT
Reverse: GGGTTTAAACGGGGCCCTTACTTGTCGTCATCGTCCTTGTAGTCGGGTGTACACACAGAACCATTACTGG
R579*
Forward: AGCGTTTAAACTTAAGATGGCTT
Reverse: GGGTTTAAACGGGGCCCTTACTTGTCGTCATCGTCCTTGTAGTCAGGACCAGGGTCCCAGTA
R1572*: The pcDNA3.1(+), ABCA1 sequence was cut with restriction enzymes BsmBI and ApaI, and the BsmBI to ApaI PCR product with a FLAG sequence was inserted just before the stop mutation of hABCA1. The primer of the inserted sequence was as follows:
Forward: TAGTTATGGGCATCTCAGAGAGACG
Reverse: GGGTTTAAACGGGGCCCTTACTTGTCGTCATCGTCCTTGTAGTCATCTGCAGAACTGTCCTTGG
The missense mutant plasmid (S1343I) was created, and the primer of the inserted sequence was as follows:
Forward: CTTCAGACACATGCCTTCAAGA
Reverse: TATCCTGCCTTCACTGGTCACA
The PCR product was cloned into ABCA1 wild type (WT), and the restriction enzyme AflII–ApaI fragment was cut out and inserted between AflII and ApaI. Then, plasmid purification was performed using NucleoSpin Plasmid EasyPure (Macherey-Nagel, Germany). Sequencing was confirmed appropriate.
U6 entry clones were generated using the BLOCK-iT U6 RNAi Entry Vector Kit (Invitrogen™ K494500). Galactocerebrosidase (GALC) knockdown plasmids were prepared as follows:
Sense target sequence: GGTTGATGAAAGAAGCTAAGA
Antisense target sequence: TCTTAGCTTCTTTCATCAACC
Double-stranded oligos were cloned into pENTER/U6 and transformed into competent cells. The sequence was confirmed to be appropriately inserted. The shRNA sequence against LacZ was used as a control. The sequences were as follows:
Sense target sequence: CTACACAAATCAGCGATTT
Antisense target sequence: AAATCGCTGATTTGTGTAG
3. Culture of HEK293T and THP-1 Cells
HEK293T cells were cultured in 4.5 g/L glucose DMEM, 100 units/mL penicillin, 100 µg/mL streptomycin sulfate, and 10% fetal bovine serum (10 µg/mL) at 37℃ in a 5% CO2 incubator (hereafter referred to as DMEM-HG).
THP-1 cells were cultured in RPMI 1640 with glutamine, 100 units/mL penicillin, 100 µg/mL streptomycin sulfate, 10% fetal bovine serum (10 µg/mL), and 50 µM 2-hydroxyethyl mercaptan at 37℃ in a 5% CO2 incubator (hereafter referred to as RPMI 1640). THP-1 cells were differentiated into macrophages using 100 nM 12-O-Tetradecanoyl Phorbol 13-Acetate, then incubated at 37℃ for 48 h. Well-differentiated macrophages were used for the experiment17-20).
4. Transfection
HEK293T cells were cultured to 80% confluence in 24-well plates. pcDNA3.1(+) 0.5 µg/well inserted with various DNAs was mixed well with Opti-MEM®I and FuGENE® HD Transfection Reagent and allowed to stand at room temperature for 10 min. Subsequently, the cells were mixed with DMEM-HG, transfected into HEK293T cells, and incubated at 37℃.
Adeno-associated virus type 2 (AAV2) expressing shRNA for LacZ or GALC under the U6 promoter was purchased from VectorBuilder (Yokohama, Kanagawa, Japan). The sequence of shRNA is identical to that described in the section on “Preparation of homemade constructs.” THP-1 cells (1.0 x 105) were seeded in a 24-well plate and differentiated with 10 nM PMA for 48 h. Thereafter, the medium was discarded, and 10 µL of the viral genomic solution at a concentration of 1.0 x 1012 GC/mL was mixed with 140 µL of the medium and applied. After overnight incubation, the medium was changed every 2 days and evaluated on day 7 21-24).
5. Establishment of Stable Cell Lines
The PiggyBac Dual promoter vector (PB510B-1, CA, USA) was cloned by inserting ABCA1_WT and S1343I between NheI and NotI. The generated plasmids were transfected into HEK293T cells, and selection was performed using 3.0 µg/mL puromycin for 2 weeks. Single colonies grown by limiting dilution were then picked up and cultured to establish stable cell lines.
6. RNA Extraction and Gene Expression Analysis
Total RNA was extracted from HEK293T cells using Sepasol-RNA I Super G (Nacalai Tesque Inc.). Reverse transcription of total RNA was performed using PrimeScriptTM RT Master Mix (Takara). The synthesized cDNA was subjected to real-time PCR (Thermal Cycler Dice Real-Time System Single, Takara) using TB Green® Premix Ex TaqTM II (Takara) to examine the expression of various genes. Using the ΔΔCt method, the expression levels of target genes were calculated as relative values, with the GAPDH value corrected as an internal standard. The primer sequences were as follows:
Gene: GALC
Forward: CGATGCATCCAACACAATCAGTATT
Reverse: TGCTGTAACTTCAACACGTCCTAAA
7. Evaluation of Expression by the Addition of Proteolysis and Protein Production Inhibitors
In the experiments, MG132 and calpeptin were used as proteolysis inhibitors25). Calpeptin, a specific calpain inhibitor, degrades ABCA1. MG132 inhibits both proteasomal and calpain degradation. The inhibition of proteolysis was considered suitable for the evaluation of protein synthesis systems.
Stable ABCA1-WT and S1343I cell lines were seeded on 12-well plates and exposed to MG132 and calpeptin at concentrations of 50 µM and 30 µg/mL for 3 h, respectively. Proteins were recovered from cultured cells26, 27). Actinomycin D and cycloheximide were used as protein production inhibitors. The concentrations of both drugs were 10 µg/mL, and proteins were collected from cultured cells after 1, 3, and 6 h of exposure28, 29).
8. Protein Extraction from the Cells
After collecting cells in lysis buffer [20 mM Tris-HCl (pH 7.9)], 100 mM NaCl, 0.5% NP-40, 1 mM EDTA [pH 8.0] mixed with cOmplete, EDTA-free diluted solution at a ratio of 25:1), the suspension was well-rotated for 30 min. The supernatant obtained via centrifugation at 15,000 rpm for 15 min was used as the protein extract.
9. Western Blotting
One-third of the total volume of 4x sodium dodecyl-sulfate (SDS) loading buffer was added to 40 µg of the protein extract and warmed at 95℃ for 5 min. Thereafter, 10% SDS polyacrylamide gel electrophoresis was performed. The separated proteins were transferred to Immobilon-P (Millipore) in a tank at 85 V for 240 min. Furthermore, the membrane was blocked with 5% skim milk (BD Biosciences) for 30 min. The primary antibody was diluted in 5% skim milk and reacted at 4℃ overnight. After washing with TBS-Tween, the secondary antibody was diluted with TBS-T and allowed to react for 30 min. Then, the membrane was reacted using the ECL Western Blotting Detection System (Amersham Pharmacia Biotech, UK), and the protein was detected using ChemiDocTM XRS Plus System (BioRad, CA, USA).
10. Efflux Assay
Forty-eight hours after transfection, the cells were mixed with DMEM+0.2% bovine serum albumin (General Grade, pH 7.0) and added with TopFluor® Cholesterol at 25 µM and incubated at 37°C for 2 h. Subsequently, the cells were washed once (1.0 mL per well) with DMEM+0.2% bovine serum albumin (general grade, pH 7.0). Next, 20 µg/mL ApoA1 was added and incubated at 37℃ overnight.
The supernatant was then collected, and the remaining cells were disrupted with 0.1 N NaOH and collected. The collected samples were seeded in 96-well plates, and fluorescence intensity was measured using a Varioskan microplate reader (Thermo Fisher Scientific). The fluorescence intensities of the supernatant and lysate are denoted by Fm and Fc. The efflux activity was calculated as follows30, 31):
Efflux (%)=[Fm / (Fm+Fc)]×100
11. Statistical Analysis
Data are expressed as means±standard error. Statistical analysis was performed using Student’s t-test to compare the two groups; p<0.05 was considered to indicate statistical significance. Bonferroni correction was applied for multiple comparisons, and analysis of variance was adopted for comparisons among three or more groups.
Results
The patients provided written informed consent, and this study was approved by the University of Tsukuba Hospital Ethics Committee with protocol numbers H26-13 in case 1 and R02-266 in case 2. In this study, two patients with marked hypo-HDL cholesterolemia who were diagnosed with Tangier disease were investigated (Table 1).
Table 1.Clinical profiles of patients with HDL deficiency on admission
|
Case 1 |
Case 2 |
| ABCA1 substitutions found (nt/aa) |
G1452A/W484*
|
C1735T/R579*
|
|
G4028T/S13431 |
C4714T/R1572*
|
| Age (years)/sex (M,F) |
20/F |
51/F |
| BMI |
26.6 |
20.1 |
| Smoking habit |
None |
None |
| Medication |
Suspended due to side effect§
|
Rosuvastatin 10 mg,
Ethyl lcosapentate 1,800 mg
|
| Total cholesterol (mg/dl) |
108 |
147 |
| HDL-cholesterol (mg/dl) |
6.2 |
3.4 |
| LDL-cholesterol (mg/dl) |
87 |
116 |
| Triglyceride (mg/dl) |
107 |
184 |
| Apo-A1 (mg/dl) |
17 |
2 |
| Atherosclerosis |
- |
+ |
| Typical TD phenotype |
+ |
+ |
| Electrocardiogram |
HR 59 bpm, RSR |
HR 70 bpm, RSR |
| ABI |
Rt. 0.99 / Lt. 1.07 |
Rt.1.08 / Lt. 1.09 |
| PWV |
Rt. 976 / Lt. 1,000 |
Rt.1228 / Lt.1238 |
| Echocardiography |
EF 53%
Mild systolic dysfunction of RV
|
EF 70%
Intermediate enlargement of LA,
Mild MR
|
| Carotid duplex ultrasonography |
|
|
| - max IMT |
Rt. 0.5 / Lt. 0.5 |
Rt. 3.2 / Lt. 3.0 |
| - Plaque Score |
0 |
11.4 |
The blood test results are from the first visit.
The physiology test results are from the most recent visit.
Abbreviations: TD, Tangier disease; RSR, Regular Sinus Rhythm ; EF, ejection fraction; IMT, intima-media thickness; RV, right ventricle; MR, mitral regurgitation
§Bezafibrate : HDL cholestrol decrease, Ezetimibe : nausea, Colestimide : fatigue
Case 1 was a 20-year-old woman with a chief complaint of gait disturbance. She had low HDL-C levels: total cholesterol, 108 mg/dL; triglyceride, 107 mg/dL; HDL-C, 6.2 mg/dL; LDL-C, 87 mg/dL; and ApoA1, 17 mg/dL. The ankle–brachial index (ABI) results were 0.99 on the right and 1.07 on the left, indicating the absence of significant lower extremity atherosclerosis. Carotid artery echocardiography revealed a mild plaque at the beginning of the right subclavian artery (max IMT 1.8 mm); however, no other obvious stenotic lesions from the common carotid artery to the internal carotid artery were observed. The patient had no previous history or family history of coronary artery disease. Tangier disease was suspected based on the presence of hand muscle weakness, decreased nerve conduction velocity, and splenomegaly. However, muscle weakness in the right upper and lower limbs also occurred; although the patient was treated as if she had a cerebral infarction, her condition did not improve. Magnetic resonance imaging (MRI) showed a cerebral white matter lesion along the left pyramidal tract (Fig.1A), which led to the suspicion of multiple sclerosis. Malignant lymphoma, sarcoidosis, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes were considered possible differentials; however, no definitive diagnosis was achieved. The patient had symptoms not suggestive of Tangier disease, such as depression, Uhthoff’s phenomenon of transient worsening of symptoms caused by heat, elevated cerebrospinal fluid proteins, and demyelinating findings on cerebral MRI. Skin biopsy revealed decreased GALC activity. Krabbe disease was also suspected. Whole-exon analysis revealed W484* nonsense mutations and S1343I missense mutation in ABCA1 (Fig.2A) as well as 12del3ins and L380F missense mutations in GALC. The patient was diagnosed with Tangier disease and Krabbe disease. The latter was considered the cause of brain-related demyelinating symptoms.
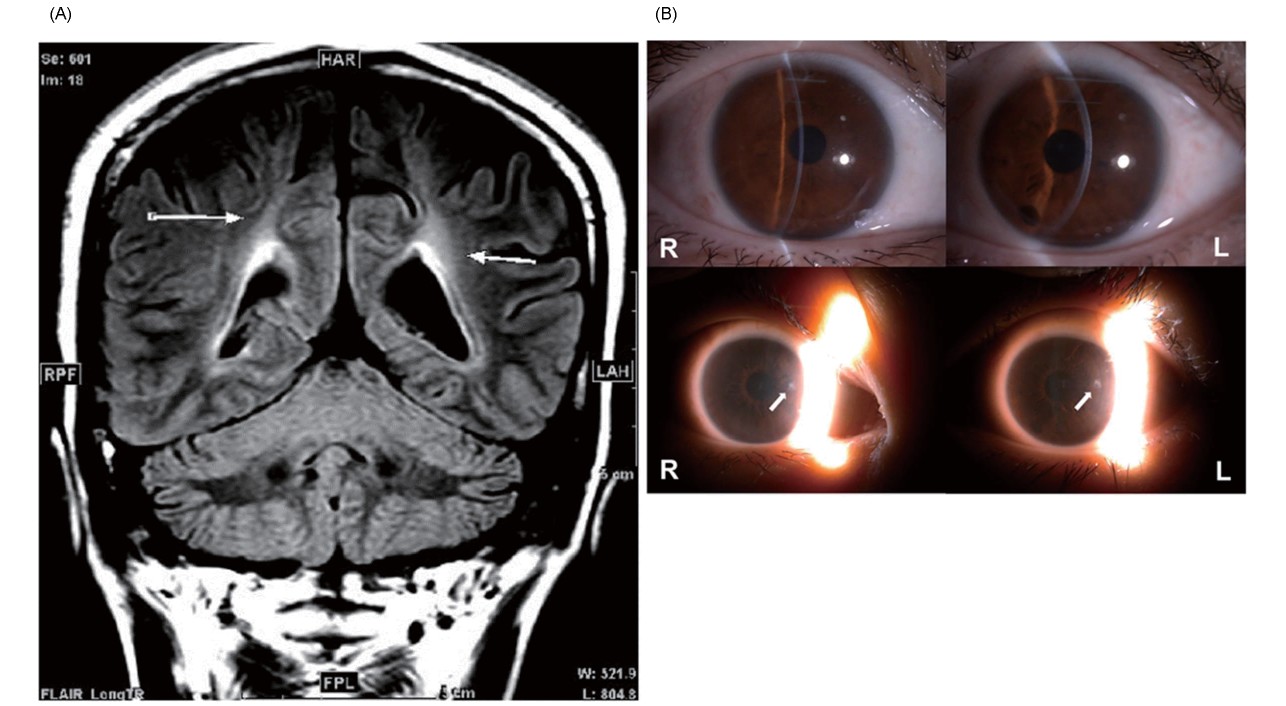

Case 2 was a 51-year-old woman with a history of tonsillectomy caused by enlarged tonsils in childhood. She started experiencing numbness in her right hand at the age of 41 years, left hand at 46, and legs at 47, and at the age of 49, she was referred to our hospital. Because of peripheral neuropathy, mild corneal opacity (Fig.1B), HDL-C of 3.4 mg/dL, and ApoA1 of 2.0 mg/dL, Tangier disease was suspected. The ABI results were 1.08 and 1.09 on the right and left, respectively, with no significant lower extremity atherosclerosis. Carotid echocardiography revealed plaques at the origin of the right subclavian artery, left common carotid artery, bilateral carotid bulbs, and bilateral internal carotid arteries, with the latter two having low echo-lucent plaques. The patient had no previous history or family history of coronary artery disease. Whole-exon analysis revealed compound heterozygosity of R579* and R1572* nonsense mutations in ABCA1 (Fig. 2B), which led to the diagnosis of Tangier disease.
The Transfected HEK293T Cells Expressed the ABCA1 Protein
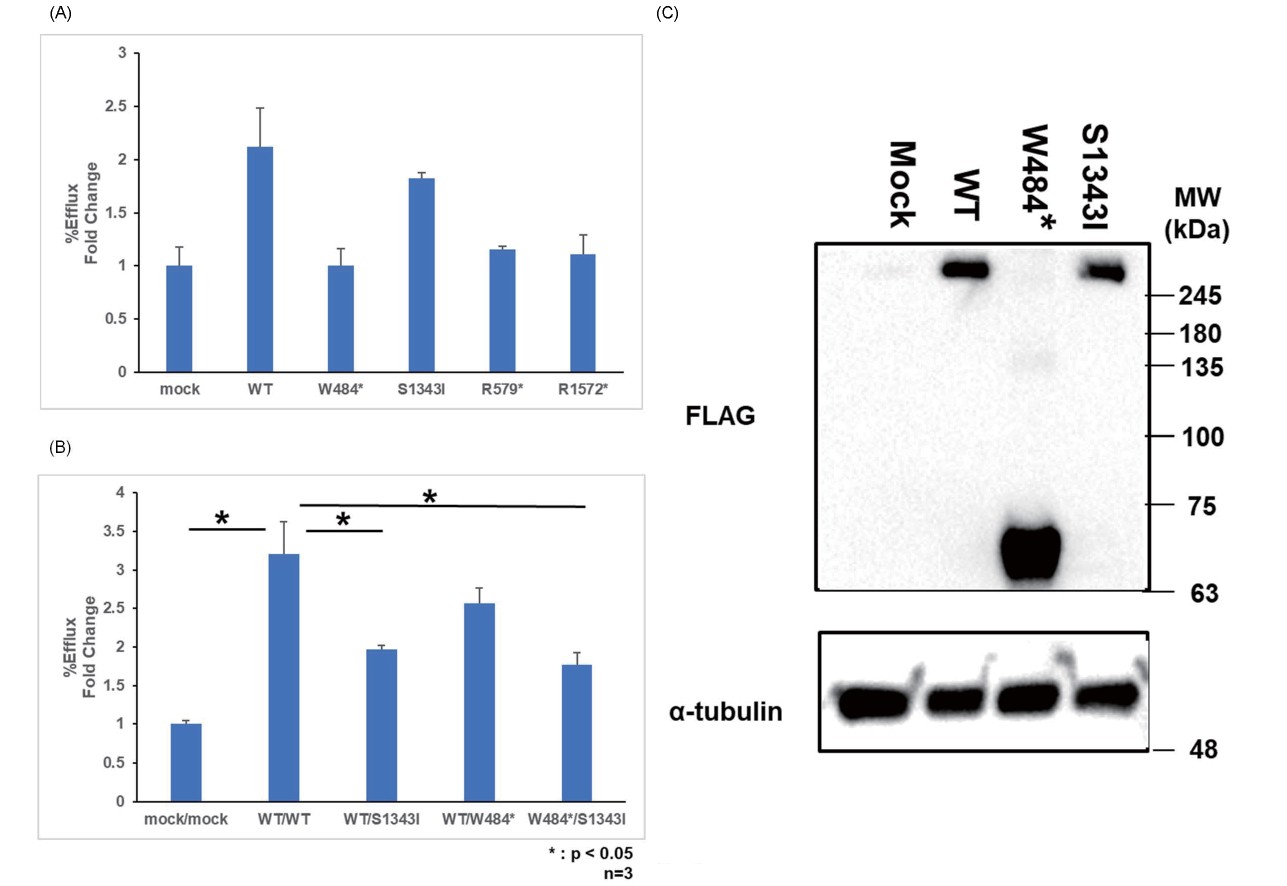
The ABCA1 protein is approximately 250 kDa in size. The ABCA1 protein in each ABCA1 mutation was evaluated by Western blotting using FLAG as the primary antibody. Furthermore, the proteins produced from ABCA1-WT and missense mutant S1343I expression plasmids were observed at the expected size of 250 kDa. In the W484* vector, which is a nonsense mutation, a plasmid with a FLAG tag was created immediately before the mutation, and the size was expected to be approximately 60 kDa. Western blotting confirmed a dark band at the expected size of 60 kDa and a stronger expression than ABCA1-WT and S1343I.
The respective R579* and R1572* nonsense mutations were also produced by creating a plasmid with a FLAG tag introduced immediately before the mutation, similar to the method employed to create the W484* mutant vector. The expected sizes of R579* and R1572* were approximately 70 and 180 kDa, respectively. Western blotting using FLAG antibodies as primary antibodies confirmed the expression of each mutant, with bands of the expected size (Fig.3).
Transient Transfection with a Mixture of Nonsense and Missense Mutant Plasmids Significantly Decreased Cholesterol Efflux Activity
We evaluated cholesterol efflux activity using the BODIPY cholesterol efflux assay in HEK293T cells transiently transfected with ABCA1-WT or a mixture of mutant plasmids.
First, ABCA1-WT, W484* nonsense mutation, and S1343I missense mutation were transiently transfected alone, and cholesterol efflux was evaluated (Fig.4A). Compared with that in ABCA1-WT, cholesterol efflux was significantly reduced by 99.9% in the W484* nonsense mutation and 26.4% in the S1343I missense mutation. Endogenous activity of the mock was excluded and calculated.
Moreover, ABCA1-WT, W484* nonsense mutation, and S1343I missense mutation were transiently transfected in a mixture of two pairs, and cholesterol efflux was evaluated. Compared with that in ABCA1-WT, cholesterol efflux was significantly reduced by 65.2% in the mixture of the W484* nonsense mutation and S1343I missense mutation (Fig.4B).
The relative expression level in the transient transfection decreased by 12.8% in the S1343I missense mutation compared with that in ABCA1-WT (Fig.4C).
The Effect of GALC Knockdown on Cholesterol Efflux in HEK293 Cell Lines Stably Expressing ABCA1 was not Observed
To evaluate protein activity, we established cell lines stably expressing the WT or S1343I-mutant ABCA1 protein, and pairs with similar expressions were selected (Nos. 1 and 4 were selected and used in subsequent experiments and denoted as PB-pCAG WT and PB-pCAG S1343I, respectively) (Fig.5A). PB-pCAG WT and PB-pCAG S1343I were evaluated in terms of cholesterol efflux, and the latter was found to have 43.1% lower efflux than the former (Fig.5B). The efflux activity of the S1343I molecule was considered reduced because the protein levels were approximately the same.

In contrast, a plasmid-knockdown GALC was created (denoted by shGALC), and gene expression reduction was confirmed via quantitative PCR and Western blotting (Fig.5C and 5D). Relative mRNA expression was 59.0% lower in shGALC. The relative protein level decreased by 81.5% in WT_shGALC. In S1343I_shGALC, the relative protein level decreased by 21.5%. This plasmid was transiently transfected into the aforementioned PB-pCAG WT and PB-pCAG S1343I, and cholesterol efflux was evaluated. Cholesterol efflux increased by 24.2% in PB-pCAG WT_shGALC. In PB-pCAG S1343I_shGALC, cholesterol efflux did not decrease. No significant decrease in efflux activity was observed (Fig.5E).
Transient Transfection of Stable Cells with a Nonsense Mutant Plasmid did not Decrease Cholesterol Efflux
Transient transfection of PB-pCAG WT and PB-pCAG S1343I stable cells with W484* nonsense mutation did not decrease cholesterol efflux. Cholesterol efflux increased by 6.1% in PB-pCAG WT_W484*. In PB-pCAG S1343I_W484*, cholesterol efflux decreased by 3.6%. Thus, the dominant-negative effect of the W484* nonsense mutation was negative (Fig.6).
The S1343I Mutation Resulted in Decreased Protein Levels Compared with that in the WT
The proteolytic and protein synthesis systems were evaluated (Fig.7A and 7B). The relative expression level of PB-pCAG_WT increased by 41.0% because of the addition of MG132. On the contrary, the relative expression level of PB-pCAG_S1343I decreased by 5.2%. By adding calpeptin, the relative expression level of PB-pCAG_WT increased by 34.4%. Conversely, the relative expression level of PB-pCAG_S1343I decreased by 9.2%. Thus, compared with that of ABCA1-WT, the addition of protease inhibitors such as MG132 and calpeptin resulted in a weaker increase in the expression of the S1343I missense mutation, suggesting that the production rate is reduced.

However, the protein expression of the S1343I missense mutation was significantly decreased by the addition of protein synthesis inhibitors, such as actinomycin D and cycloheximide, compared with that in ABCA1-WT as the elapsed time after the addition increased. The relative expression level of the S1343I missense mutation decreased by 30.9% compared with that of ABCA1-WT because of the addition of actinomycin D. By adding cycloheximide, the relative expression level of the S1343I missense mutation decreased by 44.0% compared with that of ABCA1-WT. These results suggest that the S1343I missense mutation increases the proteolysis rate (Fig.7C and 7D). Taken together, these data indicate that the S1343I missense mutation causes a decrease in protein synthesis and an increase in protein degradation, which together contribute to the decrease in protein levels.
GALC Knockdown in THP-1 Macrophages Reduced Cholesterol Efflux
Lastly, we evaluated the effect of knocking down GALC on macrophage ABCA1-mediated cholesterol efflux to clarify whether Krabbe disease affects or not. THP-1 cells were used, which contain both GALC and ABCA1 endogenously. AAV2 vectors carrying shRNA against human GALC were used to downregulate GALC expression. The relative expression levels of ABCA1 by Western blotting were reduced by 78.6% compared with the AAV shLacZ control (Fig.8A). Cholesterol efflux decreased by 19.3% compared with the AAV shLacZ control (Fig.8B). GALC hypofunction indicated a possible effect on cholesterol efflux activity in macrophages.
Discussion
In this study, the effects of genetic mutations on cholesterol efflux in patients with tangier disease were investigated.
Case 1 had a W484* nonsense mutation, which was expected to result in loss of ABCA1 function, and the cholesterol efflux of the S1343I missense mutation was also decreased relative to that of ABCA1-WT (Fig.4A). Case 2 was compound heterozygous for the R579* and R1572* nonsense mutations, which could be assumed to result in a nearly complete loss of ABCA1 function.
Although R1572* was known, W484*, S1343I, and R579* were unknown. Previous studies have reported that pathogenic ABCA1 mutations can be classified into four main groups, the majority of which have defective maturation and exhibit impaired subcellular localization. The others had impaired phospholipid translocation, defective ApoA1 binding, and abnormal ATP hydrolysis caused by mutations in the nucleotide-binding domain32).
The S1343I missense mutation in case 1 changes the hydrophilic amino acid serine to the hydrophobic amino acid isoleucine. The transmembrane domain has several hydrophobic amino acid clusters that are located next to each other, particularly those with large side chains, such as lysine, arginine, tryptophan, and tyrosine. These clusters are located near the interface where the hydrocarbon chain region changes to the aqueous phase33). The missense mutation from the hydrophilic amino acid serine to the hydrophobic amino acid isoleucine in the vicinity of the transmembrane region suggests the possibility of a functional ABCA1 mutation because of this structural mutation (Fig.9A and 9B).

PEST is a peptide sequence rich in proline, glutamic acid, serine, and threonine and is usually flanked by lysine, arginine, and histidine. The PEST sequence is located at amino acids 1283–1306 of the nucleotide-binding domain34). It is related to the proteolysis of target proteins by calpains, a subfamily of thiol proteases. ABCA1 is a target for calpain proteolysis, and the PEST sequence may help target calpain to cell surface ABCA1. ApoA1 stabilizes ABCA1 by inhibiting calpain proteolysis in a PEST sequence-dependent manner35). The S1343I missense mutation in case 1 was located near the PEST sequence, and the possibility that it affected the stability of ABCA1 protein was considered.
However, the HDL-cholesterol levels in case 1 significantly decreased to less than one-fourth of the normal levels, approximately 10 mg/dL, compared with the lower limit of 40 mg/dL, suggesting the involvement of other effects. Previously, two missense mutations, A1046D and M1091T, were found, which were next to S1343I and were heterozygous on NBD-1. However, the A1046D mutation resulted in a loss of function of approximately 59.9% of normal HDL-cholesterol levels, whereas the M1091T mutation resulted in a loss of function of only approximately 30.4%36).
ABCA1 is expressed in hepatocytes, intestinal cells, macrophages, and astrocytes and neurons in the brain37). Cholesterol synthesized in astrocytes is exported to ApoE, a major apolipoprotein in the brain, to form HDL-like particles and supply cholesterol to neurons. ABCA1 is thought to be involved in these actions.
Krabbe disease is an autosomal recessive genetic disorder resulting from a defect in GALC, which causes damage to central oligodendroglia and peripheral Schwann cells because of psychosine accumulation38). The mature form of Krabbe disease initially emerges at the age of 9 years or later with psychiatric symptoms and slowly progresses over 5–10 years with gait and cognitive and visual impairments.
Krabbe disease has been suggested to affect cholesterol efflux. It is a genetic condition that affects the lysosomal metabolism of cholesterol and galactosyl-sphingolipids. Furthermore, it is considered caused by the dysregulation of lysosomal lipid metabolism39). However, the details of the effect of Krabbe disease on cholesterol efflux remain unclear.
Case 1 had Krabbe disease in addition to Tangier disease. GALC expression in macrophages is essential in the amelioration of Krabbe disease and is important for supplying normal macrophages to Schwann cells40).
Typically, the main pathogenesis of Krabbe disease is demyelination from the accumulation of psychosine caused by GALC mutations in Schwann cells; however, secondary neuroinflammation from galactosylceramide storage in macrophages has also been reported41).
We investigated whether GALC degradation causes dysfunction of ABCA1 by suppressing GALC in cultured cells. Cholesterol efflux was not affected by GALC knockdown in PB-pCAG ABCA1-overexpressing stable cells (Fig.5E); however, in THP-1 macrophages with GALC knockdown, cholesterol efflux decreased significantly (Fig.8B). A decrease in efflux was thought to be caused by a decrease in the amount of ABCA1 protein and that GALC dysfunction due to Krabbe disease may also be involved in low HDL levels. To our knowledge, no study has reported that Krabbe disease causes low HDL. Krabbe disease may be involved in the exacerbation of hypo-HDL cholesterolemia if ABCA1 abnormality is present.
We considered the possibility that a missense mutation in ABCA1 affected protein synthesis and proteolysis systems, which caused the marked decrease in HDL-cholesterol levels in case 1. ABCA1 protein is degraded by calpain, and cysteine protease inhibitors are effective in inhibiting ABCA1 proteolysis, particularly calpain42).
Based on the above, we used MG132 as a proteasome inhibitor and calpeptin as an inhibitor of calpain and found that the S1343I mutation reduced the protein production rate (Fig.7A and 7B). Simultaneously, actinomycin D and cycloheximide were used as protein synthesis inhibitors, and the protein degradation rate due to the S1343I mutation increased (Fig.7C and 7D). This indicated that the S1343I mutation decreased protein levels and was considered a cause of the marked hypo-HDL cholesterolemia. The father of the patient in case 1 was a carrier of the W484* nonsense mutation; however, his HDL-cholesterol levels were only slightly decreased. In contrast, the mother, who had a heterozygous S1343I mutation, had normal HDL-cholesterol levels. These clinical data were not consistent with the cholesterol efflux in vitro data (Fig.4B). The efflux was not affected in WT/W484*, suggesting that this truncated protein did not have a dominant effect, whereas S1343I did. The data suggest that this point mutation could affect the stability of collateral normal protein because ABCA1 is a heterodimer protein. The involvement of this mutation in HDL-cholesterol levels was also unclear. In case 1, the patient was compound heterozygous for the W484* nonsense mutation and S1343I missense mutation from her father and mother, respectively. Furthermore, the S1343I missense mutation was thought to cause hypo-HDL cholesterolemia similar to the nonsense mutation by decreasing protein levels and molecular efflux activity.
Conclusion
In this study, we report a new pathogenic ABCA1 mutation. A heterozygous W484*/S1343I compound exhibited marked hypo-HDL cholesterolemia. In particular, the novel S1343I missense mutation, which was associated with decreased protein levels and molecular efflux activity, was thought to be similar to the nonsense mutation. A single-point mutation causes complex protein dysfunction, suggesting the unique structural specificity of ABCA1. The dysfunction of GALC also could be one of the causes.
Despite the extreme rarity, it is considered a risk factor for juvenile coronary artery disease, and differential diagnosis is necessary in cases of marked hypo-HDL cholesterolemia. The collection of such genetic information is beneficial for determining treatment strategies and genetic counseling of the proband’s offspring.
In the future, we will clarify by which missense mutations in ABCA1, such as the S1343I mutation, suppress the synthesis of ABCA1 protein and promote degradation by the calpain system and investigate the mechanism by which GALC knockdown causes the reduction in ABCA1 protein.
Acknowledgments and Notice of Grant Support
None.
Conflicts of Interest
None.
References
- 1) Bungert S, Molday LL, Molday RS. Membrane topology of the ATP binding cassette transporter ABCR and its relationship to ABC1 and related ABCA transporters: identification of N-linked glycosylation sites. J Biol Chem, 2001; 276: 23539-23546
- 2) Ye Z, Lu Y, Wu T. The impact of ATP-binding cassette transporters on metabolic diseases. Nutr Metab (Lond), 2020; 17: 61
- 3) Wang S, Smith JD. ABCA1 and nascent HDL biogenesis. Biofactors, 2014; 40: 547-554
- 4) Fitzgerald ML, Mujawar Z, Tamehiro N. ABC transporters, atherosclerosis and inflammation. Atherosclerosis, 2010; 211: 361-370
- 5) Matsuo M. ABCA1 and ABCG1 as potential therapeutic targets for the prevention of atherosclerosis. J Pharmacol Sci, 2022; 148: 197-203
- 6) Schumacher T, Benndorf RA. ABC Transport Proteins in Cardiovascular Disease-A Brief Summary. Molecules, 2017; 22: 589
- 7) Li G, Gu HM, Zhang DW. ATP-binding cassette transporters and cholesterol translocation. IUBMB Life, 2013; 65: 505-512
- 8) Rader DJ, deGoma EM. Approach to the patient with extremely low HDL-cholesterol. J Clin Endocrinol Metab, 2012; 97: 3399-3407
- 9) Kaminski WE, Piehler A, Wenzel JJ. ABC A-subfamily transporters: structure, function and disease. Biochim Biophys Acta, 2006; 1762: 510-524
- 10) Koseki M, Yamashita S, Ogura M, Ishigaki Y, Ono K, Tsukamoto K, Hori M, Matsuki K, Yokoyama S, Harada-Shiba M. Current Diagnosis and Management of Tangier Disease. J Atheroscler Thromb, 2021; 28: 802-810
- 11) Choi HY, Choi S, Iatan I, Ruel I, Genest J. Biomedical Advances in ABCA1 Transporter: From Bench to Bedside. Biomedicines, 2023; 11: 561
- 12) Bonilha I, Luchiari B, Nadruz W, Sposito AC. Very low HDL levels: clinical assessment and management. Arch Endocrinol Metab, 2023; 67: 3-18
- 13) Ogura M. HDL, cholesterol efflux, and ABCA1: Free from good and evil dualism. J Pharmacol Sci, 2022; 150: 81-89
- 14) Schaefer EJ, Anthanont P, Diffenderfer MR, Polisecki E, Asztalos BF. Diagnosis and treatment of high density lipoprotein deficiency. Prog Cardiovasc Dis, 2016; 59: 97-106
- 15) Schaefer EJ, Santos RD, Asztalos BF. Marked HDL deficiency and premature coronary heart disease. Curr Opin Lipidol, 2010; 21: 289-297
- 16) Nishida Y, Hirano K, Tsukamoto K, Nagano M, Ikegami C, Roomp K, Ishihara M, Sakane N, Zhang Z, Tsujii Ki K, Matsuyama A, Ohama T, Matsuura F, Ishigami M, Sakai N, Hiraoka H, Hattori H, Wellington C, Yoshida Y, Misugi S, Hayden MR, Egashira T, Yamashita S, Matsuzawa Y. Expression and functional analyses of novel mutations of ATP-binding cassette transporter-1 in Japanese patients with high-density lipoprotein deficiency. Biochem Biophys Res Commun, 2002; 290: 713-721
- 17) Liu Z, Thacker SG, Fernandez-Castillejo S, Neufeld EB, Remaley AT, Bittman R. Synthesis of cholesterol analogues bearing BODIPY fluorophores by Suzuki or Liebeskind-Srogl cross-coupling and evaluation of their potential for visualization of cholesterol pools. Chembiochem, 2014; 15: 2087-2096
- 18) Mao M, Lei H, Liu Q, Chen Y, Zhao L, Li Q, Luo S, Zuo Z, He Q, Huang W, Zhang N, Zhou C, Ruan XZ. Effects of miR-33a-5P on ABCA1/G1-mediated cholesterol efflux under inflammatory stress in THP-1 macrophages. PLoS One, 2014; 9: e109722
- 19) Thakkar H, Vincent V, Roy A, Singh S, Ramakrishnan L, Kalaivani M, Singh A. HDL functions and their interaction in patients with ST elevation myocardial infarction: a case control study. Lipids Health Dis, 2020; 19: 75
- 20) Thakkar H, Vincent V, Shukla S, Sra M, Kanga U, Aggarwal S, Singh A. Improvements in cholesterol efflux capacity of HDL and adiponectin contribute to mitigation in cardiovascular disease risk after bariatric surgery in a cohort with morbid obesity. Diabetol Metab Syndr, 2021; 13: 46
- 21) Yao S, Rong W, Yuan Y. Optimization of adeno-associated virus (AAV) gene delivery into human bone marrow stem cells (hBMSCs). Stem Cell Investig, 2023; 10: 3
- 22) Mori S, Takeuchi T, Kanda T. Antibody-dependent enhancement of adeno-associated virus infection of human monocytic cell lines. Virology, 2008; 375: 141-147
- 23) Zaiss AK, Cotter MJ, White LR, Clark SA, Wong NC, Holers VM, Bartlett JS, Muruve DA. Complement is an essential component of the immune response to adeno-associated virus vectors. J Virol, 2008; 82: 2727-2740
- 24) Eslami MH, Gangadharan SP, Sui X, Rhynhart KK, Snyder RO, Conte MS. Gene delivery to in situ veins: differential effects of adenovirus and adeno-associated viral vectors. J Vasc Surg, 2000; 31: 1149-1159
- 25) Zhao L, Zhao J, Zhong K, Tong A, Jia D. Targeted protein degradation: mechanisms, strategies and application. Signal Transduct Target Ther, 2022; 7: 113
- 26) Altamura C, Ivanova EA, Imbrici P, Conte E, Camerino GM, Dadali EL, Polyakov AV, Kurbatov SA, Girolamo F, Carratù MR, Desaphy JF. Pathomechanisms of a CLCN1 Mutation Found in a Russian Family Suffering From Becker’s Myotonia. Front Neurol, 2020; 11: 1019
- 27) Wang N, Chen W, Linsel-Nitschke P, Martinez LO, Agerholm-Larsen B, Silver DL, Tall AR. A PEST sequence in ABCA1 regulates degradation by calpain protease and stabilization of ABCA1 by apoA-I. J Clin Invest, 2003; 111: 99-107
- 28) Cook LB, Hinkle PM. Agonist-dependent up-regulation of thyrotrophin-releasing hormone receptor protein. Biochem J, 2004; 380: 815-821
- 29) Li J, Chen K, Dong X, Xu Y, Sun Q, Wang H, Chen Z, Liu C, Liu R, Yang Z, Mei X, Zhang R, Chang L, Tian Z, Chen J, Liang K, He C, Luo M. YTHDF1 promotes mRNA degradation via YTHDF1-AGO2 interaction and phase separation. Cell Prolif, 2022; 55: e13157
- 30) Seeree P, Janvilisri T, Kangsamaksin T, Tohtong R, Kumkate S. Downregulation of ABCA1 and ABCG1 transporters by simvastatin in cholangiocarcinoma cells. Oncol Lett, 2019; 18: 5173-5184
- 31) Adlakha YK, Khanna S, Singh R, Singh VP, Agrawal A, Saini N. Pro-apoptotic miRNA-128-2 modulates ABCA1, ABCG1 and RXRα expression and cholesterol homeostasis. Cell Death Dis, 2013; 4: e780
- 32) Qian H, Zhao X, Cao P, Lei J, Yan N, Gong X. Structure of the Human Lipid Exporter ABCA1. Cell, 2017; 169: 1228-1239.e10
- 33) Mitaku S, Sawada R, Tsuji T, Yokoyama Y. Membrane Proteins–Physics and Evolution. MEMBRANE, 2010; 35: 42-49
- 34) Oram JF, Heinecke JW. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev, 2005; 85(4): 1343-1372
- 35) Tall AR. Role of ABCA1 in cellular cholesterol efflux and reverse cholesterol transport. Arterioscler Thromb Vasc Biol, 2003; 23: 710-711
- 36) Singaraja RR, Visscher H, James ER, Chroni A, Coutinho JM, Brunham LR, Kang MH, Zannis VI, Chimini G, Hayden MR. Specific mutations in ABCA1 have discrete effects on ABCA1 function and lipid phenotypes both in vivo and in vitro. Circ Res, 2006; 99: 389-397
- 37) Jacobo-Albavera L, Domínguez-Pérez M, Medina-Leyte DJ, González-Garrido A, Villarreal-Molina T. The Role of the ATP-Binding Cassette A1 (ABCA1) in Human Disease. Int J Mol Sci, 2021; 22: 1593
- 38) Mamada N, Nakamagoe K, Shioya A, Furuta J, Sakai N, Ishii A, Tamaoka A. Adult-onset Krabbe disease presenting as acute hemiparesis and progressive demyelination detected by diffusion-weighted imaging. J Neurol Sci, 2016; 367: 326-328
- 39) Gowrishankar S, Cologna SM, Givogri MI, Bongarzone ER. Deregulation of signalling in genetic conditions affecting the lysosomal metabolism of cholesterol and galactosyl-sphingolipids. Neurobiol Dis, 2020; 146: 105142
- 40) Watanabe T. GALC Expression in Macrophages Improve Peripheral Krabbe Disease. Trends in Glycoscience and Glycotechnology, 2020; 32: E213
- 41) Weinstock NI, Shin D, Dhimal N, Hong X, Irons EE, Silvestri NJ, Reed CB, Nguyen D, Sampson O, Cheng YC, Lau JTY, Bongarzone ER, Kofler J, Escolar ML, Gelb MH, Wrabetz L, Feltri ML. Macrophages Expressing GALC Improve Peripheral Krabbe Disease by a Mechanism Independent of Cross-Correction. Neuron, 2020; 107: 65-81.e9
- 42) Yokoyama S, Arakawa R, Wu CA, Iwamoto N, Lu R, Tsujita M, Abe-Dohmae S. Calpain-mediated ABCA1 degradation: post-translational regulation of ABCA1 for HDL biogenesis. Biochim Biophys Acta, 2012; 1821: 547-551