Abstract
We constructed the four-body statistical pseudopotential proposed by Krishnamoorthy and
Tropsha, which is a type of coarse-grained potential previously used for protein-peptide
docking, using different data sets. The first data set is composed of crystal structures
of proteins that satisfy the conditions specified using PISCES, a protein sequence culling
server. The second data set consists of crystal structures of protein-protein complexes
obtained from the PDBbind-CN database. The four-body potential has 44275 patterns of
scores, depending on the type of amino acid in the quadruplet of residues and the
continuity of amino acid in the sequence of protein or peptide. While the PISCES-based
data set covers almost all patterns, docking simulations revealed that both potentials
provided comparable accuracy in reproducing experimentally determined peptide binding
poses.
1 INTRODUCTION
The COVID-19 pandemic caused by SARS-CoV-2 required rapid drug development. Docking
simulations have been used to screen drug candidates, contributing to the acceleration of
the drug development [1]. In the development of
antibody drugs, a technology that can find sequences binding predominantly to the epitope of
viral spike proteins would be useful. The docking simulation is expected to be a candidate
for such a technology.
Krishnamoorthy and Tropsha developed a four-body statistical pseudopotential for evaluating
protein stability [2]. Aita et al. applied it to the
protein-peptide docking [3]. Our group has also
developed a program for protein-peptide docking simulations using the four-body
coarse-grained potential. Unfortunately, it has become clear that the potential constructed
in the literature [2] cannot reproduce experimentally
measured binding poses of protein-peptide complexes with sufficient accuracy when used as is
[3, 4].
The four-body coarse-grained potential is statistically determined based on a data set
consisting of a large number of protein structures. Therefore, the docking result could be
strongly influenced by the contents of the data set. In this study, we aimed to improve the
accuracy of the docking simulation using the four-body statistical pseudopotential by
constructing the potentials from different data sets and comparing the docking results.
2 METHOD
2.1 Four-Body Statistical Pseudopotential
Tropsha and co-workers coarse-grained the protein structure by Delaunay tessellation as a
set of tetrahedra consisting of quadruplets, namely, representative points of four amino
acid residues [2, 5]. Each tetrahedron is classified into one of the classes α = 0, 1, 2, 3, or 4
according to the continuity in the sequence of amino acid residues (Figure 1).
The class of α = 0 means that each of the four residues in the protein/peptide sequence
is not adjacent to the others. α = 1 means that two residues are consecutive in the
sequence, and the others are separated. α = 2 is the case where two sets of two
consecutive residues are present. As for α = 3, three residues are consecutive, and the
remaining one is separated. α = 4 means that all the residues are consecutive.
Let i, j, k, and l
denote the types of amino acid residues constituting the quadruplet and α denote the
class. The score Q of the coarse-grained potential is defined by equation
(1) [2].
| Qi,j,k,lα=log[fi,j,k,lαPi,j,k,lα] | (1) |
fi,j,k,lα
is the probability that the type of amino acid residues is i,
j, k, and l when selecting one at
random from the tetrahedra of class α in the data set.
Pi,j,k,lα
is the probability that the type of amino acid residue is i,
j, k, and l and the class is α when
four amino acid residues in the data set are randomly selected. The explicit formula of
Pi,j,k,lα
is given by equation (2).
| Pi,j,k,lα=CaiajakalPα | (2) |
ai,
aj, ak, and
al are the ratio of the amino acid residues
i, j, k, and l in
the data set, respectively. Pα is the probability that the
class is α when one of the tetrahedra are randomly selected from the data set. By treating
each amino acid residue in the quadruplet as a duplicate combination, the coefficient
C is given by equation (3).
η
is the number of the types of amino acids in the quadruplet (1 ≤ η ≤ 4),
and
tiυ
is the number of type
iυ
residues in the quadruplet.
According to the literature [2], the four-body
pseudopotential was constructed using two data sets, one of which was based on a list of
protein chains from the CulledPDB directory with 2.2 Å resolution and 30% maximum sequence
identity. Another set of protein chains with a resolution no more than 2.5 Å and a maximum
sequence identity of 25% was selected from the Sequence Unique Database. The former
contained 1653 chains and the latter 967 chains.
We utilized PISCES [6, 7], which is a protein sequence culling server, to obtain a list of
protein chains at a resolution of 2.5 Å, a maximum sequence identity of 25%, and a maximum
R-value of 0.30. This resulted in a list of 10529 protein chains, based on which we
downloaded PDB files from the Protein Data Bank [8,
9] to construct the potential. Hereafter, this
data set and the corresponding four-body potential are referred to as PISCES25. The large
number of protein chains reflects the increase in the number of entries to the Protein
Data Bank during the last two decades. We prepared another data set using a database of
biomolecular complexes. 2477 structures of protein-protein complexes registered in the
PDBbind-CN database version 2020 [10] were adopted
as the components of data set. This data set and corresponding four-body potential is
referred to as PDBbind.
2.2 Docking Simulation
The PepSet Benchmark [11] was used to evaluate the
accuracy of the docking simulation. This benchmark set contains 185 protein-peptide
complexes, where peptides range in length from 5 to 20 residues. In this study, the
docking simulations of 13 complexes with 5 peptide residues were performed using our
in-house program. We excluded 5ggp, 5kds, and 6f4s because the experimental structure
shows the existence of a small molecule near the position of the peptide. An explicit
treatment of such a molecule is not subject to the protein-peptide docking based on the
four-body pseudopotential considered in this study.
In our docking simulations, peptide binding poses were searched with the protein
structures fixed. The protein surface was defined by the Defpol [12] or MSMS [13, 14] method. In the Defpol method, atomic radii of
carbon, hydrogen, nitrogen, oxygen, and sulfur were set to 1.462, 1.135, 1.392, 1.332, and
1.741 Å, respectively. The default setting of the program [14] was used for the atomic radii in the MSMS method. Density and high density
of triangulation in the MSMS method were 10.0 vertex/Å2, and the probe radius
was 2.0 Å. Search points were placed at 2.15 Å from the surface.
Proteins were coarse-grained by Delaunay tessellation and represented as a set of
tetrahedra with Cα atoms as vertices. Peptides were also treated as
coarse-grained models with Cα atoms as representative points. One of the five
amino acid residues comprising the peptide was placed at a search point, and the score of
the tetrahedra formed by the placed residue and the residues of the protein were
evaluated. The placement and evaluation were performed for all the residues. For the top
5% of search points for each residue (Residue 1 in Figure 2), the adjacent amino acid residue in the peptide sequence was placed at
a search point, and the score of the tetrahedra consisting of the placed residue and the
residues of the protein was evaluated (Residue 2 in Figure 2). For the two search points with the highest scores, the position of
the next amino acid residue was searched in the same way (Residue 3 in Figure 2). All the remaining amino acid residues are
similarly processed to obtain peptide binding poses. The search is proceeded from the
initially placed residue to the N-terminal of the peptide, and then from the remaining
residue adjacent to the initially placed one to the C-terminal.

The search for binding poses after the first residue is placed is represented as a tree
structure (Figure 2). The search does not
proceed any further except for the two nodes with the highest scores. When the low-score
nodes are removed, the tree structure becomes a binary tree, and the number of leaves is
at most 2 to the power of N − 1, where N is the number
of amino acid residues in the peptide.
For all the peptide binding poses obtained from the docking simulations, the difference
in the peptide position from the experimental structure was evaluated by Geometric center
To Geometric center Distance (GTGD) and Root Mean Squared Deviation (RMSD).
| GTGD=|1N∑i=1Nri−1N∑i=1Nriexp| | (4) |
| RMSD=1N∑i=1N|ri−riexp|2 | (5) |
N
is the number of amino acid residues in the peptide,
ri is the coordinate of the Cα atom
at the i-th amino acid residue, and
riexp
is the coordinate of the Cα atom at the i-th amino acid
residue in the experimental structure. The minimum values of GTGD determined for each
complex and those of RMSD were used as a measure of accuracy.
3 RESULTS AND DISCUSSION
3.1 Analysis of Data sets and Potential Scores
This subsection focuses on the features of the data sets used to construct the
potentials. Figure 3 shows the percentages of
amino acids counted from the data set. The differences of both data sets are small except
for alanine and serine. The overall trends with respect to the type of amino acid are
similar to the frequency of the proteome isoelectric point database consisting of about
23.9 billion amino acid residues [15]. Figure 4 shows the percentage of the tetrahedron
classes counted from the data set. The difference in appearance rates at α = 3 and α = 4
may be due to the fact that tetrahedra composed of amino acid residues between polypeptide
chains do not belong to α = 4, which means that four residues are consecutive on the amino
acid sequence of one polypeptide chain.

The score of the four-body pseudopotential expressed in equation (1) has 8855
combinations of amino acids and 5 classes, which means that there are totally 44275
patterns of i, j,
k, l, and α. We investigated the number of
combinations which are not present in the dataset (Table 1).As for the PISCES-based data set, different sequence identity
thresholds were examined. A sequence identity (%) is expressed as the number associated
with “PISCES.” Obviously, the number of non-emerging patterns correlates with the number
of data, which is the number of protein chains or that of the protein-protein complexes.
For the PISCES25 data set, the number of missing combinations was 30 out of 44275,
indicating that the data set is comprehensive. The PDBbind data set has 439 combinations
that are not present in the dataset.
Table 1. Comparison of the datasets with respect to size and non-emerging tetrahedron
patterns.
|
Number ofdata |
Number of non-emerging patterns of tetrahedra |
| PISCES30 |
14535 |
22 |
| PISCES25 |
10529 |
30 |
| PISCES20 |
6842 |
72 |
| PISCES15 |
4528 |
135 |
| PDBbind |
2477 |
439 |
Scores are not determined from equation (1) for ijklα combinations that
are not present in the data set, because
fi,j,k,lα
= 0. In such cases, a finite value of the score (−1.20) was set for the feasibility of
docking simulations. We point out the difference of the constructed potentials in the
score. They are only weakly correlated as shown in Figure 5.
3.2 Docking Simulation
Before comparing the binding poses from different potentials, we investigate whether
search points are properly placed using an algorithm that generates molecular surfaces.
The docking simulation was performed with the protein surface defined either by the Defpol
method or by the MSMS method. Table 2 lists
the number of search points and binding poses obtained from the docking simulation for
each of the 13 protein-ligand complexes.
Table 2. The number of search points and obtained binding poses.
|
Defpol |
|
MSMS |
| Searchpoint |
Binding pose |
|
Searchpoint |
Binding pose |
| 1jq8 |
3138 |
11242 |
|
2705 |
7760 |
| 1mf4 |
3221 |
11142 |
|
2769 |
9587 |
| 1nvq |
4853 |
17184 |
|
5437 |
15881 |
| 1v1t |
3829 |
14179 |
|
3769 |
13984 |
| 2aij |
5125 |
18113 |
|
5367 |
14797 |
| 2hpl |
2490 |
8823 |
|
2062 |
6930 |
| 3fp2 |
6523 |
22219 |
|
10732 |
33731 |
| 3hds |
4468 |
16045 |
|
4595 |
15003 |
| 4hdq |
5241 |
17779 |
|
6526 |
19879 |
| 4ny3 |
5362 |
19508 |
|
5871 |
21320 |
| 4×3o |
4989 |
16274 |
|
5842 |
18845 |
| 5e33 |
7630 |
24926 |
|
12119 |
35596 |
| 5onp |
5471 |
18325 |
|
6034 |
20214 |
In the case of the Defpol method, the GTGD and RMSD minima averaged over the 13 complexes
were 3.031 and 5.116 Å, respectively. Those for the MSMS method were 1.935 and 4.201 Å,
respectively. Figure 6 plots the minimum values
of GTGD and RMSD for the 13 complexes. The horizontal axis represents the case of adopting
the Defpol method to define the protein surface, and the vertical axis represents the case
of the MSMS method. The MSMS method generally provided similar or slightly smaller values
than the Defpol method, whereas the values can differ significantly. In the case of the
complex of PDBID 5e33 [16] (Figure 7), the GTGD and RMSD of the Defpol method were quite large
(> 17 Å), in contrast with the good result of the MSMS method.


All the binding poses obtained from the docking simulations of the complex with PDBID
5e33 are shown in Figure 8. The Defpol method
resulted in the peptide binding poses outside of the protein because it did not place
search points inside the protein. The MSMS method placed search points in the inner cavity
of the protein, which caused the peptide binding poses inside the protein, resulting in
qualitative agreement with the experimental structure. The difference in the ability to
place search points originates in the difference in the algorithms used to generate the
molecular surface. In the Defpol method, an ellipsoid is created outside the molecule, and
it is brought close to each atom of the protein. The MSMS method, on the other hand,
generates the surface by rolling a probe on atoms with certain radii, which is suitable
for grooves on the molecular surface and cavities inside the molecule.

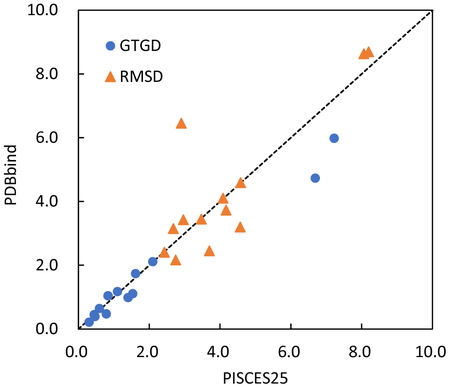
Figure 9 plots the minimum values of GTGD and
RMSD for the 13 complexes. The horizontal axis represents the case of the protein chains
based on PISCES, and the vertical axis represents the protein-protein complexes in PDBbind
as the data set used to construct the coarse-grained potential. The two and three
complexes resulted in significantly different (> 1 Å) GTGD and RMSD values,
respectively. The GTGD and RMSD minima of the potential constructed from PDBbind were
1.620 and 4.343 Å, respectively, on average for the 13 complexes, which were comparable to
the values of PISCES25 (1.935 and 4.201 Å as mentioned above).
The results presented up to here were obtained when the search of binding poses proceeds
by adopting the best and second-best cases in score when an amino acid residue is placed
at search points. The result of the search can be expressed as a binary tree. We defined
this case as N = 2 and performed the docking simulations for
N = 1 and N = 3, the latter of which corresponds to a
ternary tree. Table 3 shows the minimum values
of GTGD and RMSD averaged for the 13 complexes. As the value of N
increases, the best binding pose became closer to the experimental structure. In addition,
the accuracy in the binding pose, i.e., the smallest GTGD or RMSD was not strongly
dependent on the four-body potential.
Table 3. Averaged values of the smallest GTGDs and RMSDs (Å) from the docking simulations
employing top-
N search points.
|
Dataset |
N = 1 |
N = 2 |
N = 3 |
| GTGD |
PISCES25 |
2.343 |
1.935 |
1.526 |
|
PDBbind |
2.200 |
1.620 |
1.503 |
| RMSD |
PISCES25 |
5.027 |
4.201 |
3.770 |
|
PDBbind |
5.174 |
4.343 |
3.783 |
4 CONCLUSION
We constructed the four-body statistical pseudopotential, a type of the coarse-grained
potential previously used for protein-peptide docking. The Defpol and MSMS methods were
investigated as a technique to represent protein surfaces for the placement of search
points, with the latter providing better results. Two data sets were prepared to determine
the score of the potential. The data set obtained using PISCES is superior because of the
small number of non-emerging patterns of tetrahedra. Although the PDBbind data set was small
in amount of data, the constructed potential was comparable in docking accuracy to the
PISCES-based potential.
Acknowledgments
The docking simulations were performed using the HPC cluster of Information and Media
Center, Toyohashi University of Technology.
REFERENCES
- [1] K. Gao, R. Wang, J. Chen, L.
Cheng, J. Frishcosy, Y. Huzumi, et al., Chem. Rev., 122, 11287 (2022). ,
doi:10.1021/acs.chemrev.1c00965 PMID:35594413
- [2] B. Krishnamoorthy, A.
Tropsha, Bioinformatics, 19, 1540 (2003). , doi:10.1093/bioinformatics/btg186
PMID:12912835
- [3]T. Aita, K. Nishigaki, Y.
Husimi, Comput. Biol. Chem., 34, 53 (2010). ,
doi:10.1016/j.compbiolchem.2009.10.005
- [4]S. Matsumoto, H. Goto,
unpublished.
- [5] R. K. Singh, A. Tropsha, I.
Vaisman, J. Comput. Biol., 3, 213 (1996). , doi:10.1089/cmb.1996.3.213
PMID:8811483
- [6] G. Wang, R. L. Dunbrack,
Jr., Bioinformatics, 19, 1589 (2003). , doi:10.1093/bioinformatics/btg224
PMID:12912846
- [7] PISCES, A Protein Sequence
Culling Server,http://dunbrack.fccc.edu/pisces/.
- [8] Nucleic Acids Res., 47, D520
(2019). doi:10.1093/nar/gky949
- [9] RCSB PDB,
https://www.rcsb.org/.
- [10] PDBbind-CN database,
http://www.pdbbind.org.cn/.
- [11] G. Weng, J. Gao, Z. Wang, E.
Wang, X. Hu, X. Yao, et al., J. Chem. Theory Comput., 16, 3959 (2020). ,
doi:10.1021/acs.jctc.9b01208 PMID:32324992
- [12]C. S. Pomelli, J. Tomasi,
J. Comput. Chem., 19, 1758 (1998).
- [13]M. F. Sanner, A. J.
Olson, J. C. Spehner, Biopolymers,38, 305 (1996). ,
- [14] MSMS,
https://ccsb.scripps.edu/msms/.
- [15]L. P. Kozlowski, Nucleic
Acids Res., 50, D1535 (2022). , doi:10.1093/nar/gkab944
- [16]P. Kumar, V. Reithofer,
M. Reisinger, S. Wallner, T. Pavkov-Keller, P. Macheroux, K. Gruber, Sci. Rep., 6, 23787
(2016). , doi:10.1038/srep23787


