General Paper
Development of PolyParGen Software to Facilitate the Determination of Molecular Dynamics Simulation Parameters for Polymers
2019 Volume 5 Article ID: 2018-0034
Details
2019 Volume 5 Article ID: 2018-0034
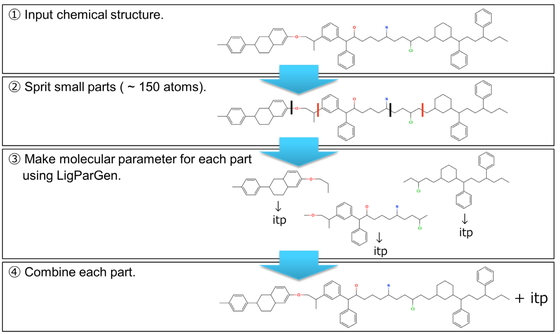
In the case that the parameters to describe the force field, such as bond angles and charges, cannot be added to the library of a molecular dynamics (MD) simulation, self-development of the force field should be considered by performing quantum mechanics calculations and/or utilizing an automatic parameter generation tool. However, these techniques are not suitable for macromolecules with a large number of atoms. Typically, the force field of an oligomer containing three unit structures (a unit at both ends and a repeating unit at the center) is calculated and converted to polymer form (both ends + central part × n). Considering this, we recently developed the program o2p, which is a semi-automated program designed to set up the force field for polymers with repeating structures. However, it is difficult to apply this method to macromolecules with complex repeating structures. Thus, in this project, we developed PolyParGen, a new open-source automatic force field generation program for Gromacs that can relatively easily and reliably simulate the MD of complex macromolecules. The proposed program (1) divides the structure of the polymer into substructures with a number of atoms within the limit of the handling size for the automatic parameter generation tool program; then, (2) acquire the parameters for each divided substructure, and finally, (3) combine the parameters of these substructures to obtain the parameters for the whole polymer. By automating these processes, it is possible to acquire a parameter of a polymer having complicated structures. This program was evaluated by simulating the polymers P3EHT and F-P3EHT in chloroform. In agreement with previous reports, fluorination was found to cause F-P3EHT to adopt an extended structure, thereby indicating the effectiveness of the proposed program.