Abstract
We had previously developed o2p, a parameter conversion semi-automation program for
polymers with repeating structures, and PolyParGen, a parameter acquisition program
primarily meant for chainlike macromolecules. These programs use the parameters obtained
via the automatic parameter generation tool. However, with these programs, it is difficult
to acquire parameters of molecules with crosslinked structures or large condensed ring
structures. To acquire these parameters, herein we improved the algorithm to develop
PolyParGen v2. Further, this program adds Antechamber as the automatic parameter
generation tool and can obtain the Amber force field parameter for polymers. This program
was evaluated via simulations using graphene, carbon nanotubes, and fullerene. Our results
agree with those of previous studies and verify the effectiveness of the proposed
program.
1 Introduction
Obtaining parameters corresponding to polymers with crosslinked structures, such as an
epoxy resin; carbon compounds such as graphene, carbon nanotubes (CNTs), fullerenes; and
derived compounds having random defects or modifications in their structures is difficult.
These compounds have been extensively studied as composite and electronic materials, and
there are many reports on Molecular Dynamics (MD) simulations [1,2,3,4,5,6,7,8,9,10], and the number is only expected to
increase in the future. Previously, we had developed o2p [11] and PolyParGen [12] to easily obtain
the files necessary for the calculation of MD for polymers with repeating structures,
randomly polymerized polymers, and randomly modified polymers. However, these programs are
difficult to apply to molecules with crosslinked structures or condensed ring structures.
Further, in the cases of materials like derivatives of fullerene, it may be impossible to
acquire parameters via LigParGen [13], even if the
number of atoms is within the limit that can be analyzed by the program. To address this
problem, we aimed to improve the algorithm of PolyParGen by dividing the molecule at the
cyclic structural sites and making it possible to obtain the files for MD simulations of
these molecules by combining the parameters of the divided molecules. This study discusses
the aforementioned improvements made to PolyParGen.
2 Development environment
Like PolyParGen, the program proposed in this paper is also a web application. We created a
user interface using PHP [14], HTML, and JavaScript,
while Python [15] was used to compose the body of the
program. In addition to LigParGen, Antechamber [16]
can also be used as an automatic parameter generation tool to obtain OPLS and Amber force
field parameters. ACPYPE [17] was used to convert the
Antechamber output to the Gromacs format. To obtain the OPLS force field, Gromacs and LAMMPS
[18] formats were used.
3 System overview
3.1 Input file format
While dividing a molecule having a conjugated double bond, such as a graphene molecule,
it is impossible to execute the division properly and add hydrogen to the terminal unless
the bonding order is accurate. Additionally, because the file input into LigParGen has no
bonding order, such as PDB format, it sometimes becomes impossible to acquire the
parameter because the automatically determined bonding order is different from the
intended bonding order. PolyParGen assumes that a molecule does not contain a large
condensed ring structure. In the case of double/triple bonds and ring structures, the
process can be carried out without any problems because the molecules are not divided, .As
these structures are also subject to division in this program, the input file format is
limited to CML to ensure the accurate acquisition of the bonding order.
3.2 Division of the molecule into partial structures
3.2.1 Selecting split atoms
As PolyParGen was primarily intended for linear polymers, the molecules were divided
along the main chain. In this program, to correspond to a spherical molecule such as
fullerene or a crosslinked structure (possibly three dimensional), the division site is
determined as follows. First, one atom is randomly selected from the input molecule
(Figure 1 (a)). Based on the selected atom,
the connected atoms are traced and added to the same group as the previous atom as a
split atom. At this time, the maximum number of atoms in one group is set so that the
whole molecule does not constitute one group. Atoms are randomly selected from the
remaining ones, and the aforementioned operation is repeated to divide the entire
molecule into split atoms (Figure 1 (b)).
Although the initial value of the maximum number of atoms is 150 (the same as the number
of atoms input to the automatic parameter generation tool), it can be set arbitrarily to
any value in the range of 1 to 150. The automatic parameter generation tool can acquire
the parameters of molecules such as fullerenes, whose parameters cannot be acquired by
intentionally dividing the molecule into small molecules even within the number of
permissible atoms corresponding to the parameter automatic generation tool.

Next, the atoms attached to the split atoms are traced, and five overlap atoms are
added. These atoms are selected to be able to maintain several uncondensed benzene
rings. Further, hydrogen (or two hydrogen atoms if it is a double bond carbon) is added
to the end depending on the bond order to form a partial molecule (Figure 1 (c)). At this time, if the number of atoms that can be
input to the parameter automatic generation tool (this time it is also set to 150) is
exceeded, the maximum number of split atoms is reduced and the process is once again
repeated from split atom creation onwards. This operation is repeated until all partial
molecules are within 150 atoms or the maximum number of split atoms is 1. Unless these
conditions are met, it might still not be possible to obtain the parameters. Further, if
one partial molecule is completely contained in another partial molecule (Figure 1 (c)), the former molecule is deleted
because it is unnecessary.
3.2.3 Maintaining the ring structure
In PolyParGen v2, the ring structure is also targeted for division, but it seems that,
in some cases, the ring structure on the polymer main chain need not be targeted for
division (Figure 2). Therefore, a large ring
structure, such as a crosslinked structure, is used as a target site for division, and
in order to suspend division at the ring structure of the polymer main chain, the size
of the ring not to be divided can be set. However, if it is set by graphene, CNTs, or
fullerenes, a divisible site cannot be found, and it becomes impossible to acquire the
parameters.
3.3 Automatic parameter generation
In this program, automatic parameter generation tools, Antechamber and LigParGen, are
executed on the same server, and the parameters of the partial structure are acquired.
Automatic parameter generation tools sometimes fail to execute the process depending on
the structure, but if it fails in this program, it is altered to try to acquire the
parameters again after further structure optimization. Although this increases the success
rate, similar to the case of PolyParGen, the parameters cannot be acquired if there is
even one partial molecule that the automatic parameter generation tool fails for, even
after structure optimization.
3.4 Combining parameters
One partial molecule is selected, and a partial molecule having a split atom which binds
to a split atom of the selected partial molecule is identified. Then, both are joined.
Further, unbound partial molecules with a split atom bonded to a split atom of a partial
molecule are retrieved and combined. This operation is repeated until all the partial
molecules are combined. Finally, the charge is adjusted in a manner similar to that in
PolyParGen.
3.5 Estimation of atomic charge
When users obtain the molecular parameters of Amber or OPLS, they have the option of
obtaining the calculated atomic charge of all split fragments via ab initio calculations,
too. The ab initio calculation software used was NWChem [19]. Users can select ESP or Mulliken atomic charges except the original atomic
charges of OPLS and Amber. Previously, we had simulated our PolyParGen using graphene
sheets [20]. We had reported good agreement of our
results with the experimental data.
4 Application examples
Based on three application examples, we evaluated the molecular parameters created by
PolyParGen. The first example comprised MD simulations of CNTs and graphene sheets in water.
CNT and graphene sheets have been reported to have a wrapped structure in water [21]. A second set of MD simulations for
[6,6]-phenyl-C61-butyric acid methyl ester (PCBM) in chlorobenzene, 1-chloronaphthalene, and
1,8-diiodooctane was also conducted. The last simulation was the estimation of the interface
energy between graphene sheet and resin. The steps for each of these simulations are given
below.
In the first example, using Avogadro [22], 6 nm ×
2 nm graphene sheets and 2-nm long CNTs (16, 0) were prepared and saved in the CML format.
The obtained CML structure file was input into PolyParGen v2, and as with the preceding
example, the itp and gro files were downloaded from this program. LigParGen was used as the
automatic parameter generation tool. The obtained MD molecular parameter files were
implemented in the 2019 version of Gromacs to perform the 10-ns MD simulation. The
simulation conditions were set to the values suggested by Gade et al. [21]. In other words, a 10 × 6 × 6 nm3 box, in which graphene
sheets and CNT were arranged at a distance of 1.4 nm, was added with 12000 water molecules
as an initial structure. The initial structures have been depicted in Figure 3 (a). As is apparent from Figure 3 (b) and (c), the MD simulation yielded a wrapped structure.

In the second example, the PCBM structure was downloaded from jasonlarkin/pcbm [23]. Structures of chlorobenzene, 1-chloronaphthalene,
and 1,8-diiodooctane were created in Avogadro. These structure files were obtained and input
into PolyParGen v2, and as with the preceding example, the itp and gro files were downloaded
from this program. PCBM yields the number of atoms that can acquire parameters via
LigParGen, but the web version of LigParGen timed out for both pdb format and mol format,
and so the parameters could not be acquired. The parameter files for chlorobenzene,
1-chloronaphthalene and 1,8-diiodooctane were separately obtained via LigParGen. In
Antechamber, an error occurred in a partial structure containing a three-membered ring
structure, and, as a result, the PCBM parameters could not be obtained. Then, ten PCBM
molecules were set in a simulation box, and none or 40 1-chloronaphthalene molecules or 40
1,8-diiodooctane molecules were inserted. Finally, 800 chlorobenzene molecules were
inserted. After NVT ensemble and NPT ensemble, under conditions of 1 atm pressure, 0.1 fs
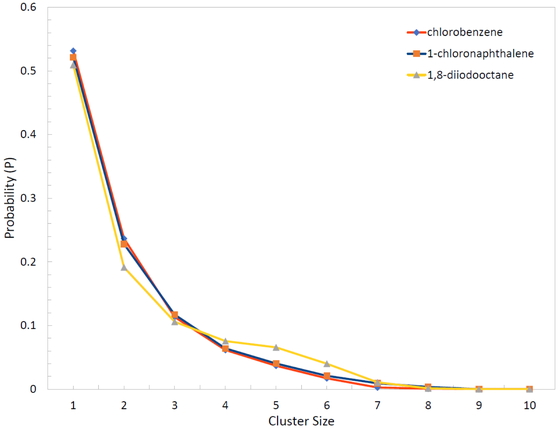
time step, 300 K temperature, 100-ns simulation was performed. The cluster size distribution
for PCBM molecules (Figure 4) confirmed that the
results agree with those previous reports [24].
In the last example, we investigated the interface energy between graphene and resin to
estimate the mechanical properties of the composite material. The simulated resins were
TriA-X, bisphenol A diglycidyl ether (DGEBA), triethylenetetramine (TETA), vinyl ester, and
polyamide 6 (PA6). Their chemical structures have been depicted in Figure 5. The simulated epoxy resin was made to react with DGEBA and
TETA. In addition, the dimensions of the graphene sheets used were 11.92 nm × 10.39 nm. The
number of carbon atoms in this graphene sheet is 4900. Three layers of graphene sheet were
set in the simulation model. To estimate the surface energy between graphene and resin, we
constructed a model in which the resins were distributed on the graphene model as depicted
in Figure 6. The interface energy is calculated by
equation (1). E denotes Energy and is calculated via molecular simulation. S denotes the
area of the graphene sheet.
E(interface
energy) =
(E(total) −
E(resin) +
E(graphene))/2S
(1)

Each interface energy has been tabulated in Table
1. The absolute values of obtained interface energies were in the order of TriA-X
polyimide> epoxy> vinyl ester> PA6. The order of the absolute values was the same
compared to that of the interfacial strengths obtained via FEM and micro bond test. Based on
the results, we could conclude that this interface energy was the one corresponding to
interfacial peeling factors caused by the inputted atomic charges and molecular parameters
of each graphene carbon atom appropriately [25].
Table 1.
The interface energy between graphene sheets and resin.
| Resin |
Interface energy[J/m2] |
Interface strength [MPa] |
| TriA-X |
−0.200 |
130 |
| Epoxy |
−0.174 |
74 |
| Vinyl ester |
−0.074 |
43 |
| PA6 |
−0.021 |
26 |
5 Conclusions
The program proposed in this paper enables us to easily prepare the files necessary for the
MD simulation of macromolecules with crosslinked and condensed ring structures. Moreover, it
makes it possible to acquire parameters for molecules for which LigParGen fails to acquire
parameters. This works even with small molecules, by dividing them into smaller molecules.
Further, even if LigParGen failed, the success rate was improved by retrying after structure
optimization. By randomizing the division of molecules, it is sometimes possible to obtain
parameters by inputting the data again into PolyParGen v2, even for molecules for which the
program once failed, and as a whole, the parameter acquisition rate was improved. However,
as with PolyParGen v2, this program is only applicable in the case of neutral molecule
correspondence, and correspondence with charged molecules is delegated to a future task.
Further, for an input molecule with a condensed aromatic ring structure, such as graphene,
five overlapping atoms are provided from the atom at the origin, and the parameters are
created by integrating those of many partial molecules. Therefore, even in the case of
condensed benzene rings, such as graphene and CNT, the carbon of the aromatic ring retains
sp2 hybridization. However, although parameters can be accurately obtained via
this method, it takes a long time to calculate them because there are many partial
molecules. For molecules in which the fused benzene ring is not as continuous as in graphene
or CNT, selecting “Maintain ring structure” can divide the molecule to protect large
aromatic rings without breaking them. If splitting is not possible while maintaining the
ring, the user is notified automatically. This program is free and open to all at the
homepage of ITOCHU Techno-Solutions Corporation, http://polypargen.com/.
Acknowledgements
This study was partially supported by JST MIRAI Grant number 19215408.
References
- [1] R. Cantrell, P. Clancy,
Surf. Sci., 602, 3499 (2008). doi:10.1016/j.susc.2008.09.027
- [2] S. R. Varanasi, O. A.
Guskova, A. John, J. U. Sommer, J. Chem. Phys., 142, 224308 (2015). ,
doi:10.1063/1.4922322 PMID:26071711
- [3] X. You, M. He, X. Cao, X.
Lyu, L. Li, Physicochem. Probl. Miner. Proces., 55, 10 (2019).
doi:10.5277/ppmp18106
- [4] B. Grosjean, M. L. Bocquet,
R. Vuilleumier, Nat. Commun., 10, 1656 (2019). , doi:10.1038/s41467-019-09708-7
PMID:30971700
- [5] Y. Wang, Y. Yang, M. Tao,
Materials (Basel), 12, 612 (2019). doi:10.3390/ma12040612
- [6] A. Gavrielides, T. Duguet,
M. Aufray, C. Lacaze-Dufaure, Int. J. Polym. Sci., 2019, 1 (2019).
doi:10.1155/2019/9604714
- [7] A. Lakshmanan, S.
Srivastava, A. Ramazani, V. Sundararaghavan, Appl. Phys. Lett., 112, 151902 (2018).
doi:10.1063/1.5022755
- [8] F. Lin, Y. Xiang, H. S.
Shen, Compos., Part B Eng., 111, 261 (2017).
doi:10.1016/j.compositesb.2016.12.004
- [9] A. Asadinezhad, P. Kelich,
Appl. Surf. Sci., 392, 981 (2017). doi:10.1016/j.apsusc.2016.09.137
- [10] A. R. Alian, M. A. N.
Dewapriya, S. A. Meguid, Mater. Des., 124, 47 (2017).
doi:10.1016/j.matdes.2017.03.052
- [11] M. Yabe, C. Sonobe, K. Ueda,
M. Takeda, J. Comput. Chem. Jpn., 16, 63 (2017).
doi:10.2477/jccj.2017-0013
- [12] M. Yabe, K. Mori, K. Ueda,
M. Takeda, J. Comput. Chem. Jpn. Int. Ed., 5, 2018-0034 (2019).
DOI:10.2477/jccjie.2018-0034
- [13] L. S. Dodda, I. Cabeza de
Vaca, J. Tirado-Rives, W. L. Jorgensen, Nucleic Acids Res., 45, W1, W331 (2017). ,
doi:10.1093/nar/gkx312 PMID:28444340
- [14] PHP (Hypertext Preprocessor),
http://www.php.net.
- [15] Python,
https://www.python.org.
- [16] J. Wang, W. Wang, P. A.
Kollman, D. A. Case, J. Mol. Graph. Model., 25, 247 (2006). ,
doi:10.1016/j.jmgm.2005.12.005 PMID:16458552
- [17] A. W. Sousa da Silva, W. F.
Vranken, BMC Res. Notes, 5, 367 (2012). , doi:10.1186/1756-0500-5-367
PMID:22824207
- [18] S. Plimpton, J. Comput.
Phys., 117, 1 (1995). doi:10.1006/jcph.1995.1039
- [19] M. Valiev, E. J. Bylaska, N.
Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T.
L. Windus, W. A. de Jong, Comput. Phys. Commun., 181, 1477 (2010).
doi:10.1016/j.cpc.2010.04.018
- [20] J. Koyanagi, N. Itano, M.
Yamamoto, K. Mori, Y. Ishida, T. Bazhirov, Adv. Compos. Mater., 28, 639 (2019).
doi:10.1080/09243046.2019.1630069
- [21] H. M. Gade, P. P. Wanjari,
S. V. V. Velpuri, Phys. Chem. Chem. Phys., 20, 22359 (2018). , doi:10.1039/C8CP02394H
PMID:30128465
- [22] Avogadro,
https://avogadro.cc/wiki/Main_Page.
- [23] jasonlarkin/pcbm,
https://github.com/jasonlarkin/pcbm.
- [24] N. R. Tummala, C. Sutton, S.
G. Aziz, M. F. Toney, C. Risko, J. L. Bredas, Chem. Mater., 27, 8261 (2015).
doi:10.1021/acs.chemmater.5b03254
- [25] S. Kasahara, J. Koyanagi, K.
Mori Makoto Yabe, The 3rd South-East Asia − Japan Conference on Composite Materials
(SEAJCCM 2019), 43 (2019)

