Relativistic DFT Calculations of Changes in NMR Chemical Shifts in Aqueous Solutions of Heavy-Metal Nitrates
2022 Volume 8 Article ID: 2021-0048
Details
2022 Volume 8 Article ID: 2021-0048
The NMR chemical shifts for metal ions in aqueous solutions of inorganic salts often change with the solution concentration. These changes are referred to herein as dilution–concentration (dc) shifts. For main-group elements, the dc shifts generally increase with increasing atomic number. We calculated the dc shifts for various metal and nonmetal ions in aqueous nitrate solutions of elements in periods 5 and 6 by the DFT method based on the spin–orbit ZORA Hamiltonian. The calculation results revealed that the dc shifts of metal and nonmetal ions with the outermost shell electron configuration of 6s2 are primarily determined by the changes in the spin–orbit interaction terms of the shielding constants. Furthermore, the spin densities of the Pb(II) ions in aqueous lead nitrate solutions under an external magnetic field were calculated using the matrix/modified Dirac–Kohn–Sham Hamiltonian, which confirmed the presence of Fermi contacts between the spin-polarized electrons and the lead nucleus. When a suitable threshold for visualizing the spin density was set, there was no Fermi contact in the infinite dilution state, whereas the upward-spin electron was in Fermi contact with the lead nucleus in the saturation state. There have been few reports of the NMR chemical shifts of heavy atoms in terms of Fermi contacts with relativistically spin-polarized electrons. We succeeded in clarifying how the dc shifts occur on the hydrated ions of heavy atoms by visualizing the Fermi contact of spin-polarized electrons with heavy-metal nuclei.
The NMR resonance lines measured for the solvated ions in aqueous solutions of various inorganic salts exhibit concentration-dependent chemical shift changes [1,2,3,4,5,6,7,8]. In this paper, we comprehensively investigate the changes in NMR chemical shifts that occur when measuring the same solvated ions at two different concentrations; we refer to such changes in the NMR chemical shifts as dilution–concentration (dc) shifts. The dc shifts between the two concentrations corresponding to infinite dilution and saturation are mainly considered in this work.
The dc shifts of metal ions generally increase with increasing atomic number. For example, the dc shifts of Pb(II) ions, where lead is a period 6 element, in aqueous nitrate solutions are larger than those of lighter metals in aqueous nitrate solutions [8]. It is likely that relativistic effects are closely related to this atomic number dependence of dc shifts.
The relativistic effects observed for the NMR resonance lines of heavy nuclei and their adjacent nuclei have been well known as “heavy-atom effects” since the early days of NMR spectroscopy. For example, normal halogen dependence (NHD) [9, 10] and inverse halogen dependence (IHD) [11] are familiar phenomena originating from such heavy-atom effects [9]. In the 1960s, Nakagawa and co-workers demonstrated using third-order Rayleigh–Schrödinger perturbation theory that the heavy-atom effects observed for the resonance lines of some alkyl halides can be ascribed to spin polarization; the resonance shifts caused by the heavy atoms were referred to as spin polarization shifts [9]. In the 1980s, four-component formalisms started to be used in theoretical calculations of the heavy-atom effects [6, 12,13,14], while Pyykkö and co-workers reported the heavy-atom effect on the heavy atom itself (HAHA effect) [6].
For the theoretical study of the dc shifts, it is important to elucidate the hydration structures of the solvated ions based on solution chemistry. Experimental results indicate that the dominant hydration structure of a solvated ion at infinite dilution is distinct from that at saturation. Nancollas demonstrated that the change in conductivity of aqueous lead nitrate solutions can be explained by rapid exchange between an aquated lead ion and a Pb(NO3)+ ion complex [15]. Recently, on the basis of this exchange equilibrium, the chemical shifts of 207Pb in aqueous lead nitrate solutions were calculated and the experimental values were accurately reproduced [10].
Pb(II) ions are known to have a hydrated structure in which the water molecules are coordinated in a hemidirected manner, which is also the case for Sn(II), Tl(I), and Bi(III) ions [16]. It has also been demonstrated that at infinite dilution the hemidirected structure of a solvated lead ion changes into a holodirected structure in which the nitrate ion and water molecules are omnidirectionally situated around the metal center [17]. The same solvation structures were applied to calculate the NMR chemical shifts for bismuth ions in aqueous solutions of bismuth nitrate [18].
In this work, to elucidate the role of relativistic effects in the dc shifts, we calculated the NMR chemical shifts of various solvated ions in aqueous solutions of nitrate salts, including the ions of both metal and nonmetal elements in periods 5 and 6. The NMR shielding constants were calculated by unrestricted density functional theory (DFT) using the spin–orbit zeroth-order regular approximation (SO-ZORA) [19,20,21] Hamiltonian to obtain the theoretical values of the dc shifts. The results revealed that for metal and nonmetal ions with the 6s orbitals as the highest occupied atomic orbitals, the dc shifts are governed by the power relationship between the paramagnetic shielding terms and the spin–orbit interaction terms.
In addition, to elucidate the relation between the spin polarization on the solvated ions and the dc shifts, we calculated and visualized the spin densities for the solvated Pb(II) ions and Cs(I) ions in the presence of an external magnetic field using a matrix/modified Dirac–Kohn–Sham (mDKS) Hamiltonian. These calculation results revealed the major factors responsible for the dc shifts of Pb(II) ions. Finally, we show that there exists a close relation between the spin–orbit interaction terms and the Fermi contact interactions of the shielding constants, which has not been reported in previous studies of the spin–orbit HAHA (SO-HAHA) effect.
In solution NMR, the resonance frequency νNMR is determined by the gyromagnetic ratio of an observed nucleus γ, the external static magnetic field B0, and the shielding constant of the nucleus σN:
| (1) |
The NMR shielding constant is defined as a tensor expressed as a 3 × 3 array based on the molecular coordinate system x, y, z. In solutions, the molecules rotate randomly, so the NMR resonance lines appear at the isotropic values
| (2) |
The shielding constant can be broken down according to the shielding mechanism as follows:
| (3) |
The Kohn–Sham DFT equation can be written as
| (4) |
| (5) |
In addition, the electron spin densities of molecules in the static magnetic field, which are strongly related to
| (6) |
| (7) |
All nitrate salts were purchased from Tokyo Chemical Industry Co., Ltd. and used without further purification. Deuterium oxide (99%) was purchased from Wako Pure Chemical Industries Ltd. Aqueous solutions of the nitrate salts were prepared in the concentration range from 0.01 to 1 mol/kg in 10% D2O, which was obtained by diluting D2O with H2O. NMR spectra were recorded at 295 K on a JEOL ECA500 spectrometer operating at 500 MHz for 1H. The π/2 pulse lengths for the observed nuclei were set to10 μs by adjusting the radio-frequency field attenuator. The recycle delays were in the range of 1–5 s. The number of co-added free induction decays varied from 8 to 2048 depending on the concentration. The NMR spectra for each nitrate salt were referenced to the lowest resonance frequency observed in the aqueous solutions.
For aqueous nitrate solutions in the infinite dilution state, hydrates in which only water molecules are bound to the central cations are dominant. In contrast, in the saturated state, the central ions of some of the hydrates are bound to nitrate ions. Hereinafter, the hydration structures corresponding to these two states will be referred to as the dilution model and the concentration model, respectively.
The initial structures of the dilution models were determined for each individual electron configuration of the outermost shells, based on previous reports [17, 29,30,31,32,33,34,35,36,37], as follows: The initial hydration structures of Rb(I) [33], Sr(II) [34], Cs(I), and Ba(II) [34] ions, in which the outermost shells of the metal ions have the s2p6 configuration, were set to square antiprismatic geometry with eight bound water molecules. The initial hydration structures of Cd(II) [31, 32], In(III) [36], Sn(IV), Sb(V), Hg(II) [35], Tl(III) [37], Pb(IV), and Bi(V) ions, in which the outermost shells of the metal ions have the s2p6d10 configuration, were set to octahedral geometry [17] with six bound water molecules. The initial hydration structure of Ag(I) ions was set to tetrahedral geometry [17, 30]; the calculation results for hydrates of Au(I) ions are not presented in this paper because no optimized hydration structure was obtained. The outermost shells of In(I), Sn(II) [17], Sb(III), Te(IV), I(V), Tl(I) [17], Pb(II) [16, 17, 29], Bi(III) [18], Po(IV), and At(V) ions have the s2 electron configuration; the initial hydration structures of Sb(III), Te(IV), I(V), Pb(II), Bi(III), Po(IV), and At(V) ions were set to hemidirected structures [17] with seven bound water molecules; the initial hydration structures of In(I), Sn(II), and Tl(I) ions were set to the structures with fourteen bound water molecules, because for In(I), Sn(II), and Tl(I) ions, the initial structures with less than fourteen water molecules did not lead to optimized structures.
To obtain the concentration models for solvated ions with the square antiprismatic geometry in their dilution models, we used the initial structure with a nitrate ion placed between the upper and lower planes of the antiprismatic structure. For solvated ions with the octahedral geometry in their dilution models, one of the water molecules was replaced by a nitrate ion to generate the initial structure for the concentration model. For the solvated ions with the hemidirected structure in their dilution models, a nitrate ion was placed around the central ion such that the initial structure for the concentration model was holodirected.
The DFT optimization of these initial structures was performed using ADF 2019 [38] with the basis functions of DZ [39] for H atoms, TZP [39] for O and N atoms, and TZ2P [39] for the central ions. For the density functional (also referred to as the exchange-and-correlation (XC) functional), we used the long-range corrected hybrid functional ωB97X [40, 41]. For the relativistic effects, the scalar relativistic effects and the spin–orbit interaction were included in the calculations by using the SO-ZORA Hamiltonian. To make the relativistic effect near the nucleus more pronounced, a Gaussian distribution model [42] was used for the nuclear charge distribution. To introduce the solvent effect, the COSMO/water scheme [43] was employed. For the structural optimization algorithm, the Hessian-based quasi-Newton-type Broyden–Fletcher–Goldfarb–Shanno (BFGS) was used. All of the optimized structures are listed in Tables S1–S46 in the Supporting Information.
The NMR shielding constants [44, 45] of the central ions in the structure-optimized hydrates in aqueous solution were determined by unrestricted DFT calculations in ADF 2019. In the NMR calculations, we used the GGA functional BP86 [46, 47] and the hybrid functionals B3LYP [48,49,50] and PBE0 [51]. Although several functionals [52] have been proposed for NMR calculations, the purpose of the present study is to investigate the effect of the solution state on the chemical shifts of heavy atoms. Therefore, rather than NMR-specific functionals, we used B3LYP and PBE0, which are widely used in general quantum chemical calculations and have been verified for accuracy. In addition, to estimate the difference between GGA functionals and the hybrid functionals, BP86 was used. The spin polarization direction due to the spin–orbit interaction was set as “non-collinear”, and gauge-independent atomic orbitals (GIAO) were adopted to avoid the problem of gauge-dependent calculation results.
The ReSpect program (version 5.1.0) [53] with the B3LYP XC functional was used to calculate the spin density. For the basis functions, we used upc-1 [54] for H atoms, dyall-vdz [55,56,57] for O and N atoms, and dyall-cvtz [55,56,57] for the central ions. Relativistic effects were introduced into the calculations at the four-component level by the mDKS Hamiltonian. A Gaussian distribution model for the nuclear charge distribution and GIAO were used in these calculations. Gauss View 6 [58] was used to visualize the calculation results.
Figure 1 presents the experimentally measured dc shifts for the metal ions in selected aqueous nitrate solutions. Although these experimental results do not precisely correspond to the calculated chemical shift differences between the dilution and concentration models, we can observe the trend in the variation of the dc shifts throughout the periodic table. Figure 1 shows that the metal ions with the s2p6 electron configuration for the outermost shell, such as Rb(I) and Cs(I) ions, exhibit small dc shifts, whereas those with electron configurations of s2p6d10, such as Cd(II) and Hg(II) ions, and s2, such as Pb(II) ions, display large dc shifts.

Experimentally measured dc shifts for metal ions in selected aqueous nitrate solutions. The chemical shifts of the metal ions were measured at several concentrations ranging from 0.01 to 1 mol/kg, and the absolute value of the maximum change in chemical shift for each ion is plotted. The horizontal axis indicates the nature of the metal ions and the period to which they belong. The expressions show the outermost shell electron configurations of the metal ions, where n denotes the period. Note that the experimental value for Hg (II) refers to mercury perchlorate rather than mercury nitrate.
Figure 2 summarizes the calculated dc shifts for metal and nonmetal ions in aqueous nitrate solutions of elements in periods 5 and 6, excluding most of the transition metals. We can see that the main features of the experimental results shown in Figure 1 are reproduced in Figure 2. There are quantitative differences between the experimental and calculated values of the dc shifts. This is because not only at 0.01 mol/kg or 1 mol/kg but also at any concentration the chemical shift is observed under equilibrium due to the rapid exchange between the complexes corresponding to the dilution and concentration models [59]. That is, the reason is that the solution states of 0.01 mol/kg and 1 mol/kg do not correspond to the dilution and concentration models, respectively.

Calculated dc shifts for metal and nonmetal ions in aqueous nitrate solutions of elements in periods 5 and 6, excluding most of the transition elements. The difference in chemical shifts between the dilution and concentration models for each ion is plotted.
In terms of functional dependence, only BP86 shows a tendency to differ significantly from the other functionals in some of the results (Figure 2). We calculated the chemical shifts of the dilution model for Pb(II) with the three functionals using tetramethyllead as a reference material, and only B3LYP and PBE0 displayed good agreement with the literature values [59]. Therefore, a functional dependence exists for BP86, and it is appropriate to base our discussion on the values obtained with B3LYP and PBE0.
We will first discuss the difference between the metal ions with the outermost shell electron configuration of s2p6, for which
The metal ions with the outermost shell electron configuration of s2p6 have
For the metal and nonmetal ions with the outermost shell electron configuration of s2p6d10 or s2, the absolute values of
The paramagnetic terms
For the metal ions with the outermost electron configuration of s2p6d10, the absolute values of
The reason why a tendency for the paramagnetic terms to have physical meaning emerges in the present analysis requires further study, and it is too early to draw any conclusions from the current data. Because the paramagnetic terms by GIAO include the contribution of the normal paramagnetic terms [60], these terms likely make the dominant contributions in the normal paramagnetic terms.
For metal ions with an outermost shell electron configuration of 6s2, changes in both the paramagnetic shielding terms and the
The
Figure 3 shows the calculated spin densities for the two models of Pb(II) ions. We found that when the threshold of the spin density visualization was set to 8.742 × 10−5 a.u. or greater, there was no Fermi contact in the dilution model, and only in the concentration model was the upward-spin electron in Fermi contact with the metal nucleus. In the concentration model, the magnetic dipole of the polarized electron spin reduces the local magnetic field of the metal nucleus more than in the dilution model. Therefore, the dc shift due to the spin–orbit interaction is negative on the δ scale. The direction of this chemical shift change is consistent with the experimental and calculated dc shifts presented in Figures. 1 and 2.
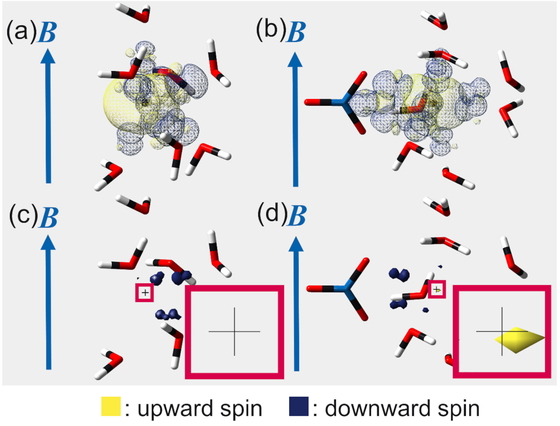
Fermi contacts between the nuclei of Pb(II) ions and spin-polarized electrons in aqueous nitrate solutions. The arrows indicate the direction of the external magnetic field. Panels (a) and (c) depict the spin density around the nucleus of a Pb(II) ion in the dilution model, while panels (b) and (d) show the spin density around the nucleus of a Pb(II) ion in the concentration model. The densities of upward and downward spins are depicted in yellow and navy, respectively. The thresholds for the spin density visualization are 1 × 10−6 a.u. in panels (a) and (b) and 8.742 × 10−5 a.u. in panels (c) and (d). In panels (c) and (d), the areas near the centers of the lead nuclei are magnified in the lower right squares. When the threshold value was less than 8.742 × 10−5 a.u., the upward spins in Fermi contact with the lead nuclei were confirmed in both the dilution and concentration models. When the threshold value was set to 8.742 × 10−5 a.u. or greater, the upward spin in Fermi contact with the lead nucleus was confirmed only in the concentration model. In the concentration model, the spin density near the center of the lead nucleus was visible until the threshold value reached 1.298 × 10−4 a.u.
For comparison, the spin densities were calculated for the dilution and concentration models of Cs(I) ions, for which the

Fermi contacts between the nuclei of Cs (I) ions and spin-polarized electrons in aqueous nitrate solutions. The arrows indicate the direction of the external magnetic field. Panels (a) and (c) depict the spin density around the nucleus of a Cs (I) ion in the dilution model, while panels (b) and (d) show the spin density around the nucleus of a Cs (I) ion in the concentration model. For the sake of clarity, the densities of the upward spins only are depicted in yellow. The thresholds for the spin density visualization are 1 × 10−6 a.u. in panels (a) and (b) and 2 × 10−6 a.u. in panels (c) and (d). In panels (c) and (d), the areas near the centers of the Cs (I) nuclei are magnified in the lower right squares. When the threshold value was greater than 2.722 × 10−5 a.u., the spin density was not observed. Because the maximum value of the threshold at which the spin density could be observed was 2.722 × 10−5 a.u., Cs (I) ions have smaller spin polarization than Pb(II) ions in the external magnetic field.
From the results of Figures 3 and 4, it is apparent that the changes in the
We have studied the effects of spin–orbit interactions on the dc shifts of metal and nonmetal ions in aqueous nitrate solutions of elements belonging to periods 5 and 6, primarily through quantum chemical calculations. For the solvated ions with outermost shell electron configurations of s2p6, s2p6d10, and s2, the dc shifts increased in that order for a given period, which was confirmed by both calculations and experiments.
We further investigated the components of the NMR shielding constants through calculations using the SO-ZORA Hamiltonian. It was found that for metal ions with an outermost shell electron configuration of s2p6d10, the dc shifts were determined by the changes in the dominant paramagnetic shielding terms, whereas for metal and nonmetal ions with an outermost shell electron configuration of s2, the dc shifts were determined by the changes in both the paramagnetic shielding and spin–orbit interaction terms.
We also calculated the spin densities, which are induced by both the external magnetic field and spin–orbit interactions, for the dilution and concentration models of selected metal ions using the mDKS Hamiltonian. These calculations were applied to examine the spin-polarized electrons in Fermi contact with the ionic nuclei. These calculations ultimately revealed distinct changes in the spin densities on the nuclei of Pb(II) ions with the outermost shell electron configuration of s2 between the dilution and concentration models. These results provide a clear explanation for one of the origins of the changes in the spin–orbit interaction terms in the shielding constants calculated using the SO-ZORA Hamiltonian.
In previous studies of the SO-HALA effects, it has been pointed out that changes in the molecular structure lead to changes in the
Optimized structures for various solvated ions in aqueous nitrate solutions.
We are grateful to Dr. Ayaki Sunaga for helpful discussions and comments on the manuscript. We gratefully acknowledge the work of past members of our laboratory, Takumi Yokoyama, Tatsuki Sato, and Kenta Tasaki. We would also like to thank the ReSpect program team for kindly providing the ReSpect program, especially Dr. Michal Repisky for responding to us via email.