Abstract
Apple latent spherical virus (ALSV) vectors have been shown to effectively induce stable virus-induced gene silencing (VIGS) in a wide range of plant species, including rosaceous fruit tree species such as apple (Malus × domestica Borkh.), pear (Pyrus communis L.), and Japanese pear (P. pyrifolia Nakai). In this study, we attempted to develop a VIGS-based gene evaluation system for two Prunus fruit tree species, apricot and Japanese apricot, using ALSV vectors. A partial sequence of the P. armeniaca PHYTOENE DESATURASE (ParPDS) gene was cloned and ligated into the T-DNA region of a binary vector, pBICAL2, designed based on RNA2 of ALSV. The resultant pBICAL2-ParPDS was introduced into a disarmed Agrobacterium strain, EHA105. pBICAL1, a binary plasmid for the expression of ALSV RNA1 in plants, was also introduced into EHA105. Leaves of Nicotiana benthamiana were infected with pBICAL1/EHA105 and pBICAL2-ParPDS/EHA105 simultaneously to produce and amplify recombinant ALSV particles. The amplified ParPDS-ALSV in N. benthamiana was isolated and infected into the cotyledons of apricot and Japanese apricot seedlings by particle bombardment. Although our attempts to infect wild and recombinant ALSVs into Japanese apricot seedlings were unsuccessful, uniform discoloration of the upper leaves, a typical phenotype of PDS knock down, was observed several weeks after inoculation in apricot seedlings. We discuss the possible use of this VIGS-based gene evaluation system in Prunus.
Introduction
Plants have developed several defense mechanisms against small infectious agents such as viruses and viroids. RNA silencing is a well-known defense mechanism of plants and prevents virus systemic infection. This plant intrinsic defense mechanism detects and cleaves double-stranded RNA that is formed during virus replication and/or hairpin structures of viral RNA. Cleavage of double-stranded RNA into small pieces by Dicer-like (DCL) enzymes generates viral small interfering RNA (siRNA). The siRNA thus produced is amplified by RNA-dependent RNA polymerase (RdRP) and DCL enzymes, and integrated into the RNA-induced silencing complex (RISC). The RISC serves as a guide to induce the degradation of complementary viral RNA systemically (Mine and Okuno, 2008; Vance and Vaucheret, 2001; Voinnet, 2005; Waterhouse et al., 2001).
Virus-induced gene silencing (VIGS) is a plant gene analysis tool that was developed from the RNA-based defense mechanism of plants against viruses. VIGS is triggered by the infection of viral vectors carrying partial sequences of endogenous plant genes. Plant mRNA corresponding to the partial sequence carried by the viral vector is degraded in a homology-dependent manner. VIGS is a very useful tool for the functional analysis of genes in plants because knock-down phenotypes of VIGS target genes can be observed in a relatively short period, several weeks after inoculation of viral vectors, without the lengthy transformation steps usually necessary for the Agrobacterium-mediated stable transformation system (Lu et al., 2003; Purkayastha and Dasgupta, 2009; Senthil-Kumar and Mysore, 2011; Waterhouse and Helliwell, 2003). Moreover, this method has the potential to silence multicopy genes or genes of different plant species with similar genetic backgrounds (Purkayastha and Dasgupta, 2009). Because of these advantages, several types of viruses have been modified into VIGS vectors and used for the silencing of endogenous genes in herbaceous model plants and crops such as Arabidopsis thaliana, Nicotiana benthamiana, Solanum species, legume species, rice (Oryza sativa L.), wheat (Triticum aestivum L.), barley (Hordeum vulgare L.), maize (Zea mays L.), cotton (Gossypium hirsutum L.), and strawberry (Fragaria × ananassa) (Bennypaul et al., 2012; Brigneti et al., 2004; Chai et al., 2011; Constantin et al., 2004; Ding et al., 2006; Faivre-Rampant et al., 2004; Fu et al., 2005; Holzberg et al., 2002; Turnage et al., 2002; Tuttle et al., 2008; Yan et al., 2012; Zhang and Ghabrial, 2006). In contrast, only a few studies on virus vectors for VIGS in fruit tree species had been reported until recently, mainly because of the lack of efficient viral vectors. Furthermore, woody plants are usually more resistant to virus infections and the mechanical inoculation methods routinely used for herbaceous plants are not readily applicable to fruit tree species (Yamagishi et al., 2010; Yanase et al., 1979).
Recently, apple latent spherical virus (ALSV) vectors have been shown to effectively induce stable VIGS of endogenous genes in a wide range of plant species including not only herbaceous plant species but also rosaceous fruit tree species, such as apple (Malus × domestica Borkh.), pear (Pyrus communis L.), and Japanese pear (P. pyrifolia Nakai) (Igarashi et al., 2009; Sasaki et al., 2011; Yamagishi and Yoshikawa, 2009). ALSV, which is classified into the genus Cheravirus, consists of isometric virus particles ca. 25 nm in diameter, and contains two ssRNA species (RNA1 and RNA2) and three capsid proteins (Vp25, Vp20, and Vp24) (Li et al., 2000). Although there have been no reports so far of the application of the ALSV vector system to Prunus, it should be able to be used for VIGS in Prunus because the broad host range of ALSV includes the subtribe Pyrinae of the subfamily Spiraeoideae to which Prunus belongs (Potter et al., 2007). Furthermore, ALSV infects the subfamily Rosoideae in the Rosaceae (Ito et al., 2012).
Prunus includes many economically important fruit and nut tree species such as peach (P. persica Batsch), apricot, Japanese apricot, sweet cherry (P. avium L.), European plum (P. domestica L.), Japanese plum (P. salicina Lindl.), and almond [P. dulcis (Mill.) D. A. Webb.]. Characterization of the functions of genes associated with agriculturally important traits such as flowering, dormancy, self-incompatibility, fruit quality, disease resistance, abiotic tolerance, and various other developmental processes is very important to promote Prunus breeding. However, Prunus is among the most recalcitrant species to Agrobacterium-mediated transformation. There have been only a few reports describing successful transformations in Prunus (Gao et al., 2010; Pérez-Clemente et al., 2004; Petri et al., 2008; Ramesh et al., 2006; Wang et al., 2011). As it is currently difficult to evaluate gene functions in Prunus, an ALSV-mediated gene evaluation system might provide an alternative tool to enable such studies in Prunus.
In this study, we investigated whether a VIGS-based gene evaluation system using ALSV vectors could be applied to two Prunus fruit tree species, apricot and Japanese apricot. ALSV could infect apricot by particle bombardment. Furthermore, recombinant ALSV could induce VIGS of the endogenous PDS gene with no obvious viral symptoms. However, ALSV infection into Japanese apricot using the same method was unsuccessful. The possible use of the ALSV-mediated VIGS system for gene function analysis in Prunus species is discussed.
Materials and Methods
Plant materials and RNA extraction
Seedlings of apricot ‘Heiwa’ and Japanese apricot ‘Nanko’ were grown in a growth chamber under LD conditions (16/8 h light/dark photoperiod) at 25°C. Young leaves of the apricot and Japanese apricot seedlings were collected, immediately frozen in liquid nitrogen, and stored at −80°C until use. Total RNA was extracted from the collected leaves using the hexadecyl-trimethyl-ammonium bromide (CTAB) method and cDNA was synthesized as described in Yamane et al. (2011). Seeds of apricot and Japanese apricot were rinsed under running tap water overnight, placed on a layer of filter paper moistened with distilled water in petri dishes, and germinated under 4°C, dark conditions. Just after germination, the seedlings were used for viral inoculation as described below.
Construction of ALSV vectors
We selected the PDS gene as a target for VIGS because it was one of the most widely used markers of VIGS in previous studies (Kumagai et al., 1995). Based on nucleotide sequence alignment of PDS genes of apricot (DDBJ/EMBL/GenBank Acc. No. AY822065) and other woody plant species, a conserved region of the apricot PDS gene (ParPDS; 108 bp; Fig. 1A) was amplified from cDNA from apricot leaves using the primer pair ParPDS-F and ParPDS-R (Table 1). The RT-PCR products were cloned into the pGEM-T easy vector (Promega, Madison, WI, USA) and their DNA sequences were determined using a CEQ8000 and DTCS Quick Start Master Mix (Beckman Coulter, Fullerton, CA, USA).

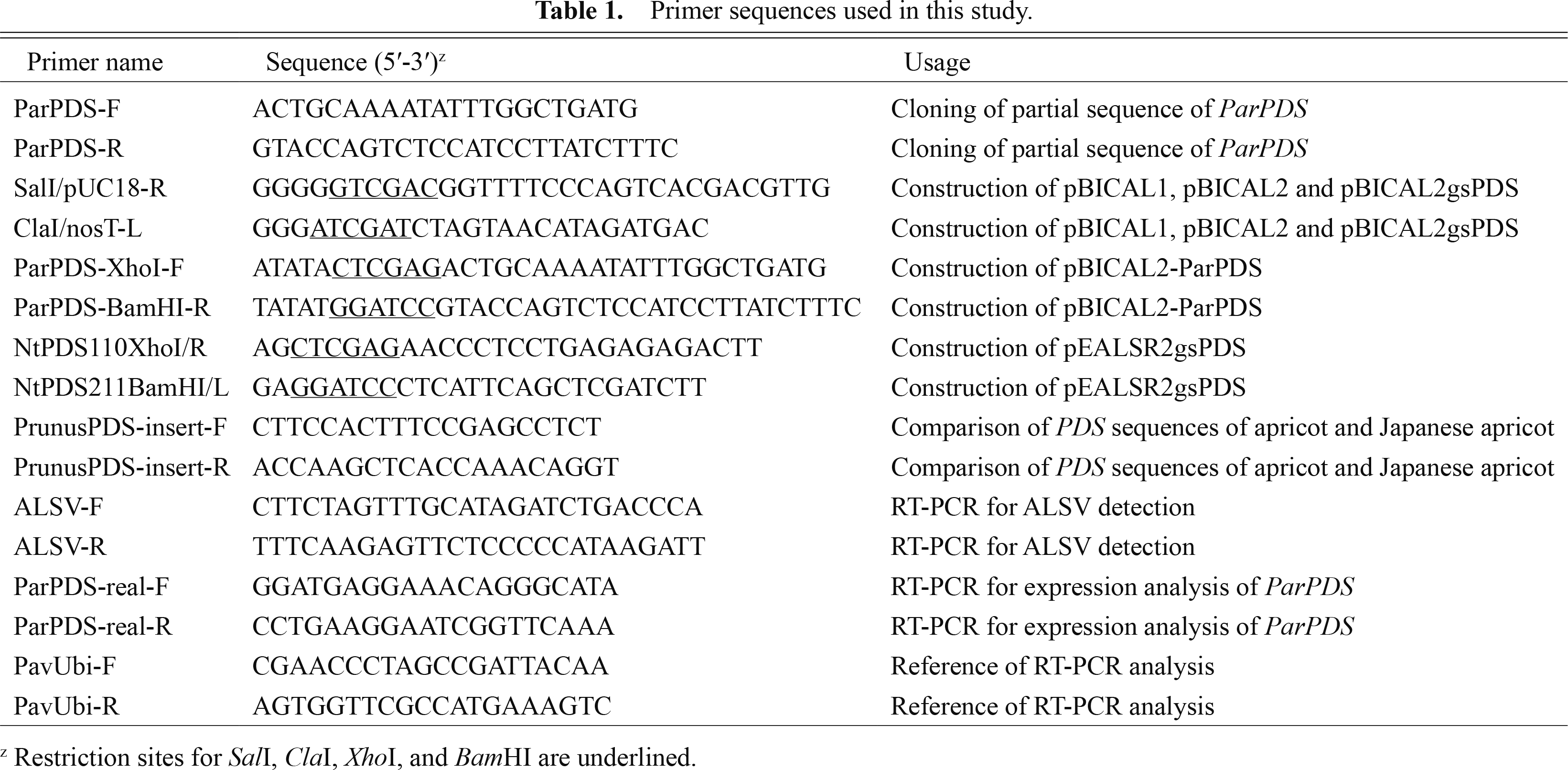
To construct pBICAL1 (Fig. 2), an 8-kb fragment containing ALSV RNA1 expression cassette was amplified from pEALSR1 (Li et al., 2004) using primers SalI/pUC18-R and ClaI/nosT-L, which included SalI and ClaI sites, respectively (Table 1). The generated PCR product was digested with SalI and ClaI and inserted into the same sites of pBICP35 (Mori et al., 1991), producing pBICAL1. For pBICAL2 construction (Fig. 2), a 4.5-kb fragment containing ALSV RNA2 expression cassette was amplified from pEALSR2L5R5 (Li et al., 2004) using primers SalI/pUC18-R and ClaI/nosT-L (Table 1). The generated PCR product was digested with SalI and ClaI and inserted into the same sites of pBICP35, producing pBICAL2.
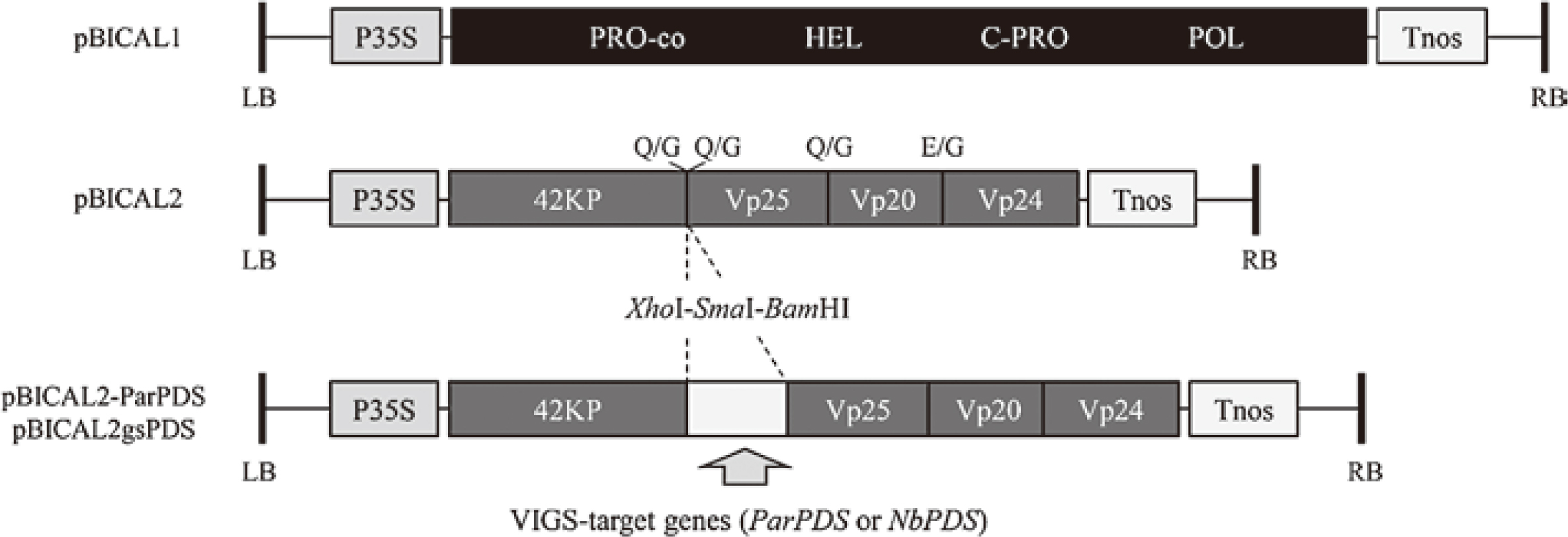
To construct ALSV vectors carrying the partial sequence of ParPDS, the cloned DNA sequence was amplified by PCR with PrimeSTAR GXL DNA polymerase (Takara Bio, Shiga, Japan) using the primer pair ParPDS-XhoI-F and ParPDS-BamHI-R, which included XhoI and BamHI sites, respectively (Table 1). The PCR products were double-digested with XhoI and BamHI and ligated in frame with the coding sequences for the movement and Vp25 capsid proteins flanking the cloning site of pBICAL2 digested with the same enzymes. The resultant construct, designated pBICAL2-ParPDS (Fig. 2), was introduced into a disarmed Agrobacterium strain, EHA105, by electroporation using electrocompetent EHA105 cells produced by a standard technique using 10% (w/v) glycerol and a MicroPulser electroporator (BIO-RAD, Hercules, CA, USA). pBICAL1 and pBICAL2 were also introduced separately into EHA105.
To construct ALSV vectors carrying the partial sequence of the N. benthamiana PDS (NbPDS) gene, 102-nucleotide partial fragment of NbPDS (Fig. 1B) was amplified from cDNA derived from N. benthamiana RNA using primers NtPDS110XhoI/R and NtPDS211BamHI/L, which included XhoI and BamHI sites, respectively (Table 1). The fragment was digested with XhoI and BamHI and inserted into the same sites of pEAL2SR2L5R5, producing pEALSR2gsPDS. A 4.6 kb-fragment containing ALSV RNA2 expression cassette was amplified from pEALSR2gsPDS using primers SalI/pUC18-R and ClaI/nosT-L (Table 1). The generated PCR product was digested with SalI and ClaI and inserted into the same sites of pBICP35. The resultant construct, designated pBICAL2gsPDS (Fig. 2), was introduced into EHA105.
Comparison of PDS sequences of apricot and Japanese apricot
It has been reported that more than 23 successive nucleotides with 100% sequence identity to the target gene are necessary for VIGS (Purkayastha and Dasgupta, 2009). As apricot and Japanese apricot have similar genetic backgrounds, ALSV vectors for VIGS of ParPDS could be applicable for the silencing of Japanese apricot PDS (PmPDS). To confirm this, we determined the nucleotide sequence of the PmPDS region corresponding to the partial sequence of ParPDS used for the construction of pBICAL2-ParPDS. RT-PCR was performed with cDNA from Japanese apricot leaves using the primer pair PrunusPDS-insert-F and PrunusPDS-insert-R (Table 1). PCR amplification was performed with a program of 95°C for 3 min followed by 35 cycles of 95°C for 20 sec, 60°C for 15 sec, and 72°C for 45 sec. After alkaline phosphatase and exonuclease treatment, direct sequencing of the PCR products was performed using a CEQ8000 and DTCS Quick Start Master Mix (Beckman Coulter).
Amplification of ALSV vectors in N. benthamiana
N. benthamiana plants were used to produce and amplify wild-type (wt) and recombinant ALSV particles. They were grown under 16 h LD conditions at 25°C for about 10 days after sowing, and then grown under 16 h LD conditions at 23°C. Young seedlings 17–21 days after germination (3–4 leaf stage; Fig. 3A) were used for Agrobacterium-mediated viral inoculation. Leaves of N. benthamiana were simultaneously inoculated with pBICAL1/EHA105 and pBICAL2/EHA105, or pBICAL1/EHA105 and pBICAL2-ParPDS/EHA105, using toothpick. Frozen stocks of EHA105 harboring each plasmid were added separately to about 200 μL LB liquid medium. The suspensions were incubated for 2–3 h at 28°C and spread on LB agar plates with an appropriate antibiotic. After incubation for 1–2 days at 28°C, fresh colonies of pBICAL1/EHA105 and pBICAL2/EHA105, or pBICAL1/EHA105 and pBICAL2-ParPDS/EHA105, were suspended together in 100 μL of 10 mM MgCl2 containing acetosyringone (100 μg·mL−1). The suspensions were kept at 23°C for 3–4 h. The tip of a sterilized toothpick was dipped in the suspension and inserted into N. benthamiana leaves. Four to six inoculations were made at different points on each leaf of 17–21 days old young N. benthamiana plants (Fig. 3A). The inoculated plants were grown under 16 h LD conditions at 23°C. pBICAL1/EHA105 and pBICAL2gsPDS/EHA105 were also infected into N. benthamiana because chlorosis of upper leaves, a phenotype of PDS gene silencing caused by the amplification of NbPDS-ALSV, could be used as a good indicator to estimate the movement and amplification of wtALSV and ParPDS-ALSV.

Successful viral infection was determined by the microtissue direct RT-PCR method 2–3 weeks post inoculation (wpi) according to Hosokawa et al. (2006) with slight modifications. PCR amplification was performed with a program of 32 cycles of 95°C for 20 sec, 58°C for 15 sec, and 72°C for 45 sec with an initial heating at 95°C for 3 min using the primer pair ALSV-F and ALSV-R (Table 1) corresponding to nucleotide positions 1396–1421 and 1573–1598 of ALSV RNA2 (AB030941), respectively. This primer pair was designed to amplify the region containing the cloning site to confirm the existence of inserted sequences in ALSV RNA2. Young upper leaves and stipules of infected N. benthamiana were collected, stored at −80°C, and used for the extraction of total RNA to be used as an inoculum.
Viral inoculation to apricot and Japanese apricot seedlings
Total RNA containing amplified wtALSV or ParPDSALSV was isolated from infected N. benthamiana using TRIzol reagent (Life Technologies Japan, Tokyo, Japan), purified by phenol/chloroform extraction, and used for inoculation of apricot and Japanese apricot seedlings by particle bombardment, essentially as described in Yamagishi et al. (2010). Briefly, 8 mg gold particles (0.6 μm in diameter; BIO-RAD) were sonicated in 50 μL RNase-free water for at least 1 min using Ultrasonic Cleaner SU-27TH (Sibata Scientific Technology, Tokyo, Japan). Then, 50–100 μL of RNase-free water containing 200 μg total RNA extracted from infected N. benthamiana was added to the gold particle suspension with continuous vortex mixing. After mixing, a 1/10 volume of 5 M ammonium acetate and twice the volume of isopropyl alcohol was added to precipitate the RNA. The mixture was kept at −20°C for at least 1 h. After removing the supernatant, the gold particles coated with total RNA were washed with 1 mL dehydrated 99.5% ethanol three times to remove water, and resuspended in 2.4 mL dehydrated 99.5% ethanol. The suspension was used to coat the inside of a plastic tube (50 cm) with gold particles according to the manufacturer’s instructions. The gold-coated tubing was cut into pieces (1.25 cm), each of which contained 5 μg total RNA, and used for biolistic inoculation.
The seed coats were gently removed from apricot and Japanese apricot seedlings just after germination and the cotyledons were bombarded (Fig. 4A, B) with gold particles coated with total RNA at a pressure of 280–300 psi using a Helios Gene Gun system (BIO-RAD). Bombardments were performed four and three times on each side of the cotyledons of apricot and Japanese apricot, respectively. After acclimatization in the dark at 4°C and 100% relative humidity, the inoculated seedlings were planted in soil and grown in a growth chamber under 16 h LD conditions at 25°C.
RT-PCR analysis
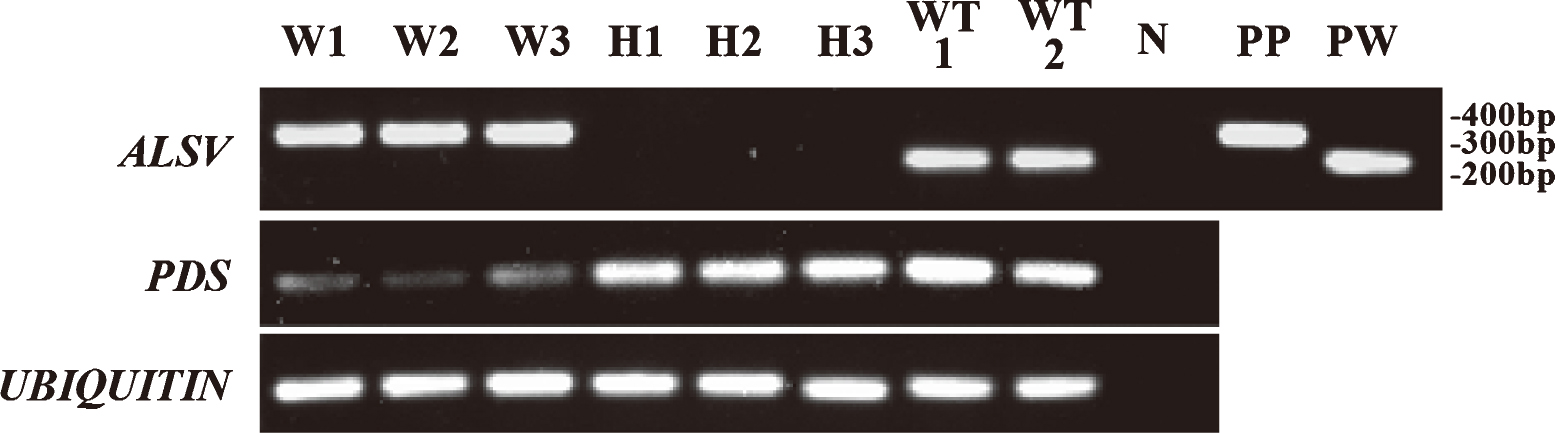
Total RNA was extracted from the upper leaves of inoculated apricot and Japanese apricot seedlings by a small-scale CTAB method according to Sasaki et al. (2011) with slight modifications. cDNA was synthesized using 500 ng total RNA with ReverTra Ace qPCR RT Master Mix with gDNA Remover and random primers (TOYOBO, Osaka, Japan). cDNA equivalent to the amount synthesized from 5 ng total RNA was used as a template for RT-PCR analysis. RT-PCR for detection of ALSV was carried out using the same primer pair and PCR program that were used for virus detection in N. benthamiana. Semi-quantitative RT-PCR analysis of mRNA levels of the PDS gene was performed using the primer pair ParPDS-real-F and ParPDS-real-R (Table 1) and a PCR program consisting of 32 cycles of 95°C for 20 sec, 60°C for 15 sec, and 72°C for 45 sec with initial heating at 95°C for 3 min. The PCR products were electrophoresed on a 1% (w/v) agarose gel, stained with ethidium bromide, and detected using a UV illuminator. In addition, real-time RT-PCR with the same primer pair was conducted using SYBR Green Master Mix and a LightCycler 480 (F. Hoffmann-La Roche AG, Basel, Switzerland). This PCR was performed with a program of 95°C for 5 min followed by 45 cycles of 95°C for 10 sec, 60°C for 20 sec, and 72°C for 1 sec. Gene-specific amplification was confirmed by melting curve analysis. Three technical replicates were performed. As a reference, the UBIQUITIN gene-specific primer pair PavUbi-F and PavUbi-R (Yooyongwech et al., 2008; AF298826; Table 1) was used for RT-PCR and real-time RT-PCR analyses.
Tissue blot analysis
The distribution of ALSV in the leaves of inoculated N. benthamiana and apricot seedlings was confirmed by tissue blot analysis as described previously (Yamagishi et al., 2009) using a DIG-labeled RNA probe complementary to ALSV RNA2.
Results and Discussion
Efficient amplification of ALSV vectors in N. benthamiana
In the original method (Li et al., 2004), ALSV vectors for particle bombardment were produced and amplified in Chenopodium quinoa by mechanically inoculating infectious cDNA clones designed from ALSV RNA1 and RNA2. In this study, we used N. benthamiana to amplify ALSV vectors more efficiently. The use of N. benthamiana made it possible to avoid the time-consuming inoculation step that was necessary for the original viral amplification method with C. quinoa. Using modified binary Ti-plasmids with CaMV35S promoter sequence (Fig. 2), agroinfection became possible. When Agrobacterium tumefaciens strains pBICAL1/EHA105 and pBICAL2gsPDS/EHA105 were inoculated using a toothpick, a uniform photo-bleached phenotype, which is a typical symptom of VIGS of the NbPDS gene, was observed in the upper leaves of almost all inoculated plants 2–3 wpi, as described previously (Igarashi et al., 2009; Fig. 3A, B). Using this phenotypic change as an indicator of viral amplification, direct microtissue RT-PCR and tissue blot analyses were performed to confirm the systemic infection of wtALSV or ParPDS-ALSV in N. benthamiana inoculated with pBICAL1/EHA105 and pBICAL2/EHA105, or pBICAL1/EHA105 and pBICAL2-ParPDS/EHA105. When RT-PCR of the upper leaves was conducted using ALSV RNA2-specific primers, almost all inoculated plants produced 251-bp and 353-bp PCR fragments corresponding to wtALSV and ParPDS-ALSV, respectively (data not shown). Although Igarashi et al. (2009) indicated that ALSV vectors carrying a partial sequence of the PDS gene longer than 300 bp lost their insert sequences 30–40 days post inoculation (dpi) in tobacco plants, such loss of inserted sequences was not observed in our experiments for N. benthamiana plants infected with NbPDS-ALSV and ParPDS-ALSV. The shorter length of partial PDS gene sequences used in this study (102 bp of NbPDS and 108 bp of ParPDS) could explain the different results obtained in our study and by Igarashi et al. (2009). Tissue blot analysis of inoculated plants using a DIG-labeled RNA probe complementary to ALSV RNA2 indicated that ALSV was accumulated uniformly in the upper leaves (Fig. 3C, D). These results suggested that N. benthamiana was successfully infected with ALSV vectors by Agrobacterium-mediated viral inoculation using a toothpick, which could offer an easy and fast way to produce and amplify ALSV vectors for particle bombardment.
Infection of apricot and Japanese apricot seedlings with ALSV vectors
The infection rates of ALSV for apple, pear, Japanese pear, and soybean [Glycine max (L.) Merr.] were reported to be almost 100% when ALSV RNAs were introduced into the cotyledons of seedlings just after germination by particle bombardment (Sasaki et al., 2011; Yamagishi and Yoshikawa, 2009; Yamagishi et al., 2010). As very high infection efficiencies were obtained with apple, pear, and Japanese pear, we followed these methods to inoculate apricot and Japanese apricot. Total RNA isolated from N. benthamiana infected with wtALSV and ParPDS-ALSV was used to coat gold particles, and the cotyledons of apricot and Japanese apricot seedlings just after germination were bombarded with these gold particles using a helium gun (Fig. 4A, B). RT-PCR with ALSV RNA2-specific primers produced PCR fragments corresponding to wtALSV (251 bp) and ParPDS-ALSV (353 bp) for the upper leaves 2–3 wpi in 2 of 12 seedlings infected with wtALSV (16.7%) and in 5 of 45 seedlings infected with ParPDS-ALSV (11.1%), respectively (Table 2; Fig. 5). Apricot seedlings infected with wtALSV exhibited no obvious virus infection symptoms and showed normal growth comparable to healthy plants with no viral infection (Fig. 4D, E). These results suggested that ALSV vectors could be effectively used to evaluate gene functions in apricot because viral symptoms often hinder the correct evaluation of phenotypic changes caused by silencing of the VIGS-target gene. Conversely, we detected neither wtALSV nor ParPDS-ALSV from the leaves of 10 and 25 Japanese apricot seedlings infected with these virus vectors, respectively (Table 2). This result may indicate that species-or cultivar-dependent ALSV susceptibility differences exist in Prunus. As we only used seedlings from a single cultivar each of apricot and Japanese apricot, it is unclear whether the differential susceptibility was dependent on species or cultivar. In either case, we need to consider genotype-dependent variation in ALSV susceptibility when the ALSV vector system is used to evaluate gene functions in Prunus.

The PDS gene encodes a key enzyme in the carotenoid biosynthesis pathway and carotenoid pigments are essential for the protection of chlorophyll from photo-oxidation (Bartley and Scolnik, 1995). Because inhibition of the PDS gene leads to rapid degradation of chlorophyll and results in a photo-bleached phenotype in plants, it is widely used as a visible marker of VIGS (Kumagai et al., 1995).
Sequence analysis revealed that the partial sequence of ParPDS inserted into the ALSV construct contained more than 23 successive nucleotides with 100% identity to PmPDS (Fig. 1C). Thus, we targeted the PDS gene for VIGS in apricot and Japanese apricot using the same ALSV vector, ParPDS-ALSV.
Of the 45 apricot and 25 Japanese apricot seedlings bombarded with ParPDS-ALSV, 5 apricot and 0 Japanese apricot seedlings were successfully infected with ParPDS-ALSV (Table 2). Uniform discoloration of the upper leaves, a typical phenotype of PDS gene knock down, was observed in all infected apricot seedlings 2–3 wpi (5–6 leaf stage; Fig. 4D). The discoloration first appeared along the veins of the upper leaves (Fig. 4C) and then spread to the whole area of newly generated leaves (Fig. 4D). Tissue blot analysis was performed to confirm the systemic infection of ALSV in these photo-bleached leaves because RT-PCR analysis could check virus infection of only a small part of leaves. The result showed that ALSV was accumulated uniformly in silenced upper leaves (Fig. 6).

The infected seedlings exhibited slightly stunted growth, which could have been caused by the inhibition of photosynthesis, and stopped growing within 3 months post inoculation (16–18 leaf stage). The silenced phenotype persisted throughout plant growth. However, photo-bleached unfolded upper leaves occasionally turned yellow or light green later in their development (data not shown). Similar results were observed in rose, in which PDS-silenced photo-bleached leaves turned green, and then newly developed leaves showed a photo-bleached phenotype (Ito et al., 2012). Although ALSV vectors containing the insert sequence were detected by RT-PCR from leaves showing this “recovery phenotype” (data not shown), the efficiency of VIGS may have declined because of reduced virus concentration or other environmental factors. In fact, it has been reported in previous studies that some growth conditions such as temperature or humidity affect the efficiency or stability of silencing (Chellappan et al., 2005; Fu et al., 2006; Tuttle et al., 2008).
We further investigated the amount of PDS-mRNA in the leaves of apricot seedlings infected with ParPDSALSV by semi-quantitative RT-PCR and real-time RT-PCR analysis to confirm that the discoloration of leaves was caused by silencing of the PDS gene. Semi-quantitative RT-PCR indicated that the amount of PDS transcripts in the photo-bleached leaves of apricot seedlings infected with ParPDS-ALSV was considerably decreased compared with the seedlings infected with wtALSV or healthy, uninoculated seedlings (Fig. 5). Real-time RT-PCR also indicated that the amount of PDS-mRNA was distinctly reduced in silenced white leaves (Fig. 7). These results collectively suggested that ALSV vectors could successfully induce VIGS of the PDS gene. ALSV-mediated VIGS should therefore be a useful tool for the evaluation of endogenous gene functions in apricot.

The results obtained in this study, however, raise several issues that need to be addressed before gene functions in apricot and other Prunus fruit tree species can be effectively assessed using ALSV-mediated VIGS. First, the infectivity of ALSV could vary with species or cultivar in Prunus. We need to select the right species or genotypes for ALSV-mediated gene functional analysis. Second, the efficiency of ALSV infection into apricot using particle bombardment was low compared with ALSV infection into apples and pears (Sasaki et al., 2011; Yamagishi et al., 2010). Although this could be also related to genotype differences, we need to optimize infection methods and plant growth conditions after inoculation. Third, the stability of the virus or VIGS has yet to be determined fully. Finally and most importantly, we also need to develop an ALSV infection system for adult plant materials so that we can test gene functions in reproductive organs. Further studies are needed before we can fully utilize the VIGS-based gene evaluation system for Prunus using ALSV vectors.
Literature Cited
- Bartley, G. E. and P. A. Scolnik. 1995. Plant carotenoids: pigments for photoprotection, visual attraction, and human health. Plant Cell 7: 1027–1038.
- Bennypaul, H. S., J. S. Mutti, S. Rustgi, N. Kumar, P. A. Okubara and K. S. Gill. 2012. Virus-induced gene silencing (VIGS) of genes expressed in root, leaf, and meiotic tissues of wheat. Funct. Integr. Genomics 12: 143–156.
- Brigneti, G., A. M. Martin-Hernández, H. Jin, J. Chen, D. C. Baulcombe, B. Baker and J. D. G. Jones. 2004. Virus-induced gene silencing in Solanum species. Plant J. 39: 264–272.
- Chai, Y. M., H. F. Jia, C. L. Li, Q. H. Dong and Y. Y. Shen. 2011. FaPYR1 is involved in strawberry fruit ripening. J. Exp. Bot. 62: 5079–5089.
- Chellappan, P., R. Vanitharani, F. Ogbe and C. Fauquet. 2005. Effect of temperature on geminivirus-induced RNA silencing in plants. Plant Physiol. 138: 1828–1841.
- Constantin, G. D., B. N. Krath, S. A. MacFarlane, M. Nicolaisen, I. E. Johansen and O. S. Lund. 2004. Virus-induced gene silencing as a tool for functional genomics in a legume species. Plant J. 40: 622–631.
- Ding, X. S., W. L. Schneider, S. R. Chaluvadi, M. A. Rouf Mian and R. S. Nelson. 2006. Characterization of a Brome mosaic virus strain and its use as a vector for gene silencing in monocotyledonous hosts. Mol. Plant Microbe Interact. 19: 1229–1239.
- Faivre-Rampant, O., E. M. Gilroy, K. Hrubikova, I. Hein, S. Millam, G. J. Loake, P. Birch, M. Taylor and C. Lacomme. 2004. Potato virus X-induced gene silencing in leaves and tubers of potato. Plant Physiol. 134: 1308–1316.
- Fu, D. Q., B. Z. Zhu, H. L. Zhu, W. B. Jiang and Y. B. Luo. 2005. Virus-induced gene silencing in tomato fruit. Plant J. 43: 299–308.
- Fu, D. Q., B. Z. Zhu, H. L. Zhu, H. X. Zhang, Y. H. Xie, W. B. Jiang, X. D. Zhao and Y. B. Luo. 2006. Enhancement of virus-induced gene silencing in tomato by low temperature and low humidity. Mol. Cells 21: 153–160.
- Gao, M., M. Kawabe, T. Tsukamoto, H. Hanada and R. Tao. 2010. Somatic embryogenesis and Agrobacterium-mediated transformation of Japanese apricot (Prunus mume) using immature cotyledons. Sci. Hortic. 124: 360–367.
- Holzberg, S., P. Brosio, C. Gross and G. P. Pogue. 2002. Barley stripe mosaic virus-induced gene silencing in a monocot plant. Plant J. 30: 315–327.
- Hosokawa, M., Y. Matsushita, H. Uchida and S. Yazawa. 2006. Direct RT-PCR method for detecting two chrysanthemum viroids using minimal amounts of plant tissue. J. Virol. Meth. 131: 28–33.
- Igarashi, A., K. Yamagata, T. Sugai, Y. Takahashi, E. Sugawara, A. Tamura, H. Yaegashi, N. Yamagishi, T. Takahashi, M. Isogai, H. Takahashi and N. Yoshikawa. 2009. Apple latent spherical virus vectors for reliable and effective virus-induced gene silencing among a broad range of plants including tobacco, tomato, Arabidopsis thaliana, cucurbits, and legumes. Virology 386: 407–416.
- Ito, H., M. Ochiai, H. Kato, K. Shiratake, D. Takemoto, S. Otagaki and S. Matsumoto. 2012. Rose phytoene desaturase gene silencing by apple latent spherical virus vectors. HortScience 47: 1278–1282.
- Kumagai, M. H., J. Donson, G. Della-Cioppa, D. Harvey, K. Hanley and L. K. Grill. 1995. Cytoplasmic inhibition of carotenoid biosynthesis with virus-derived RNA. Proc. Natl. Acad. Sci. USA 92: 1679–1683.
- Li, C., N. Sasaki, M. Isogai and N. Yoshikawa. 2004. Stable expression of foreign proteins in herbaceous and apple plants using Apple latent spherical virus RNA2 vectors. Arch. Virol. 149: 1541–1558.
- Li, C., N. Yoshikawa, T. Takahashi, T. Ito, K. Yoshida and H. Koganezawa. 2000. Nucleotide sequence and genome organization of Apple latent spherical virus: a new virus classified into the family Comoviridae. J. Gen. Virol. 81: 541–547.
- Lu, R., A. M. Martin-Hernandez, J. R. Peart, I. Malcuit and D. C. Baulcombe. 2003. Virus-induced gene silencing in plants. Methods 30: 296–303.
- Mine, A. and T. Okuno. 2008. Viruses and RNA scilencing. Virus (Japan) 58: 61–68 (In Japanese with English abstract).
- Mori, M., K. Mise, K. Kobayashi, T. Okuno and I. Furusawa. 1991. Infectivity of plasmids containing brome mosaic virus cDNA linked to the cauliflower mosaic virus 35S RNA promoter. J. Gen. Virol. 72: 243–246.
- Pérez-Clemente, R. M., A. Pérez-Sanjuán, L. García-Férriz, J. P. Beltrán and L. A. Cañas. 2004. Transgenic peach plants (Prunus persica L.) produced by genetic transformation of embryo sections using the green fluorescent protein (GFP) as an in vivo marker. Mol. Breed. 14: 419–427.
- Petri, C., K. Webb, J. M. Hily, C. Dardick and R. Scorza. 2008. High transformation efficiency in plum (Prunus domestica L.): a new tool for functional genomics studies in Prunus spp. Mol. Breed. 22: 581–591.
- Potter, D., T. Eriksson, R. C. Evans, S. Oh, J. E. E. Smedmark, D. R. Morgan, M. Kerr, K. R. Robertson, M. Arsenault, T. A. Dickinson and C. S. Campbell. 2007. Phylogeny and classification of Rosaceae. Plant Syst. Evol. 266: 5–43.
- Purkayastha, A. and I. Dasgupta. 2009. Virus-induced gene silencing: A versatile tool for discovery of gene function in plants. Plant Physiol. Biochem. 47: 967–976.
- Ramesh, S. A., B. N. Kaiser, T. Franks, G. Collins and M. Sedgley. 2006. Improved methods in Agrobacterium–mediated transformation of almond using positive (mannose/pmi) or negative (kanamycin resistance) selection-based protocols. Plant Cell Rep. 25: 821–828.
- Sasaki, S., N. Yamagishi and N. Yoshikawa. 2011. Efficient virus-induced gene silencing in apple, pear and Japanese pear using Apple latent spherical virus vectors. Plant Methods 7: 15.
- Senthil-Kumar, M. and K. S. Mysore. 2011. New dimensions for VIGS in plant functional genomics. Trends Plant Sci. 16: 656–665.
- Turnage, M. A., N. Muangsan, C. G. Peele and D. Robertson. 2002. Geminivirus-based vectors for gene silencing in Arabidopsis. Plant J. 30: 107–117.
- Tuttle, J. R., A. M. Idris, J. K. Brown, C. H. Haigler and D. Robertson. 2008. Geminivirus-mediated gene silencing from Cotton leaf crumple virus is enhanced by low temperature in cotton. Plant Physiol. 148: 41–50.
- Vance, V. and H. Vaucheret. 2001. RNA silencing in plants—defense and counterdefense. Science 292: 2277–2280.
- Voinnet, O. 2005. Induction and suppression of RNA silencing: insights from viral infections. Nat. Rev. Genet. 6: 206–220.
- Wang, H., N. Alburquerque, L. Burgos and C. Petri. 2011. Adventitious shoot regeneration from hypocotyl slices of mature apricot (Prunus armeniaca L.) seeds: A feasible alternative for apricot genetic engineering. Sci. Hortic. 128: 457–464.
- Waterhouse, P. M. and C. A. Helliwell. 2003. Exploring plant genomes by RNA-induced gene silencing. Nat. Rev. Genet. 4: 29–38.
- Waterhouse, P. M., M. B. Wang and T. Lough. 2001. Gene silencing as an adaptive defense against viruses. Nature 411: 834–842.
- Yamagishi, N. and N. Yoshikawa. 2009. Virus-induced gene silencing in soybean seeds and the emergence stage of soybean plants with Apple latent spherical virus vectors. Plant Mol. Biol. 71: 15–24.
- Yamagishi, N., S. Sasaki and N. Yoshikawa. 2010. Highly efficient inoculation method of apple viruses to apple seedlings. Julius-Kuhn-Archiv. 427: 226–229.
- Yamane, H., T. Ooka, H. Jotatsu, Y. Hosaka, R. Sasaki and R. Tao. 2011. Expressional regulation of PpDAM5 and PpDAM6, peach (Prunus persica) dormancy-associated MADS-box genes, by low temperature and dormancy-breaking reagent treatment. J. Exp. Bot. 62: 3481–3488.
- Yan, H. X., D. Q. Fu, B. Z. Zhu, H. P. Liu, X. Y. Shen and Y. B. Luo. 2012. Sprout-vaccum-infiltration: a simple and efficient agroinoculation method for virus-induced gene silencing in diverse solanaceous species. Plant Cell Rep. 31: 1713–1722.
- Yanase, H., A. Yamaguchi, G. I. Mink and K. Sawamura. 1979. Back transmission of apple chlorotic leafspot virus (type strain) to apple and production of apple topworking disease symptoms in Maruba Kaido (Malus prunifolia Borkh. var. ringo Asami) Ann. Phytopath. Soc. Japan 45: 369–374.
- Yooyongwech, S., A. K. Horigane, M. Yoshida, M. Yamaguchi, Y. Sekozawa, S. Sugaya and H. Gemma. 2008. Changes in aquaporin gene expression and magnetic resonance imaging of water status in peach tree flower buds during dormancy. Physiol. Plant. 134: 522–533.
- Zhang, C. and S. A. Ghabrial. 2006. Development of bean pod mottle virus-based vectors for stable protein expression and sequence-specific virus-induced gene silencing in soybean. Virology 344: 401–411.