Abstract
骨格系の組織のうち,骨・筋肉組織は,人体の支持組織として不可欠な組織である.この筋骨格系の維持機能は非常に重要である.組織が損傷する,すなわち骨折,筋肉打撲といった筋骨格系の組織損傷は日常しばしば発生し,その組織損傷は直ちに正しく修復されなければならない.この修復機構は厳密に体内で制御されている.組織損傷の下では,凝固線溶系の活性化が不可欠である.組織内血管が,筋骨格系組織の損傷によって,破綻がおき,瞬く間に凝固線溶系カスケードが作用し修復機能が働く.フィブリンが形成され止血に働き,役割を終えたフィブリン網はプラスミンによって分解される.このプラスミンを中心とした線溶系は,実はフィブリンの溶解といった古典的作用だけでなく,多様な生理的および病態生理的作用を果たしている.本稿では,この凝固線溶機能と骨と筋肉組織修復の関与とその機序に関して,今までの知見とともに概説したい.
1.はじめに
人体の組織のうち,骨,筋肉といった筋骨格系は,内臓臓器を守り,また日常動作を可能にしている必須な臓器である.骨はカルシウムの貯蔵組織として胎生期から種々のサイトカインやホルモンにより時間的・空間的に厳密に制御され一生涯にわたり恒常性が保たれる.筋は体の40%を占めるといわれ,伸張と収縮により関節の可動を可能にしている.この筋骨格系は日々の動作により,ときとして組織損傷を受ける.幸いなことに,通常我々の体は,この損傷に素早く反応し,修復するといった治癒反応が起きている.組織治癒としては,出血,止血,急性炎症,組織増殖,リモデリングを経る.しかしながら,炎症疾患である糖尿病や肥満,自己免疫疾患といった疾患を有する場合,この修復機構が損なわれ,骨修復不全である偽関節や骨量減少であるオステオペニア,病的な筋量減少であるサルコペニアといった状態に陥る1).さらに組織損傷の修復の失敗は重大な障害を招く.骨癒合不全や筋肉治癒障害によって四肢機能低下や,慢性的な疼痛,可動域の低下,四肢の機能障害等をもたらし,ADLを著しく損なう結果を招く2, 3).
凝固線溶機能は人体維持にとって不可欠な機能であり,筋骨格においても非常に重要な役割を果たしている.骨折など外傷や,術中術後に伴う凝固能力の低下は過剰出血を引き起こすことがあり,ときに致死的となる.一方,人工関節術後や脊椎手術後の凝固過剰は血栓を引き起こすことがあり,時として重篤な症状を引き起こす.生体内において,この凝固線溶機能のバランスが厳密に制御され,筋骨格組織が維持されている2, 4).
筋骨格系システムは豊富なエネルギー供給が必要とされ,そのためにはエネルギー源を運搬する血行が豊富でなければならず,筋骨格系も血管が張り巡らされている.それゆえ,筋骨格組織損傷による血管の損傷は避けられない.血管の損傷により,出血,その後血腫すなわち血腫凝固となり,組織修復と共にそれは消失する.凝固線溶系システムは,出血に伴うフィブリンによる凝固と,血腫を溶解するプラスミンによる線溶と機能しているが,このシステムは,組織損傷後の一次止血といった古典的役割に寄与しているだけでなく,組織修復の中で様々に寄与していることが示されている5–10).線溶系タンパクであるプラスミンは,マトリックスプロテアーゼ(MMP)の活性化を介して細胞外基質を分解し,これによる細胞外基質に存在する血管内皮細胞増殖因子(vascular endothelial growth factor: VEGF)やトランスフォーミング成長因子(transforming growth factor-β: TGF-β)などの成長因子を遊離させる11–13).また単核球やマクロファージの移動を促進したり14),細胞内シグナルに関しては,細胞膜に発現するuPA受容体やプロテアーゼ活性化受容体(protease-activated receptors: PARs)を介するシグナルの活性化など多岐にわたる15).このようにプラスミンは古典的なフィブリンの溶解といった作用だけでなく,血管新生,走化性,恒常性維持や細胞分化等組織修復の際の様々な現象に寄与している.ここでは筋骨格損傷と凝固線溶系の関係を我々の研究成果を中心にこれまでの知見とともに説明する.
2.線溶系と筋骨格系損傷後の急性反応
線溶系タンパクであるプラスミンは,組織型プラスミノゲンアクチベータ(tPA)及びウロキナーゼ型プラスミノゲンアクチベータ(uPA)により前駆体であるプラスミノゲンが分解され,酵素活性を有するタンパクである.プラスミンの古典的作用以外の多岐にわたる生体での機能ゆえに,組織損傷後のプラスミン活性は非常に重要で,これは厳密に制御されなければならない.
ひとたび筋骨格系損傷が発生すると,急性の反応が起きる.すなわち,出血の制御,組織の低酸素化の制御といった反応である.まず凝固系が活性化し,止血に作用する.この際,フィブリンが血管内,血管外に沈着する.血管内フィブリンは血栓となり血液漏出を防ぐ.一方血管外フィブリンは損傷組織内で細胞外基質マトリックスとなり,組織修復を保護する.すなわちフィブリンマトリックスにおいて,tPAやuPAによりプラスミノゲンからプラスミンに分解され活性化し,フィブリンを分解し,低酸素状態を改善,組織機能を回復させる.プラスミンは沈着フィブリンを分解し,さらに炎症反応を促進させ,損傷した壊死組織などの除去に作用する.それらが完了したのち,プラスミンはVEGFの産生を促進させ,血管新生を導くことにより,局所の低酸素状態を改善する12).このように効果的な組織修復は,フィブリン形成から惹起されるプラスミン活性といったイベントが厳格に制御されてはじめて完成する.いいかえれば,このバランスが崩れると筋骨格系損傷後,組織修復の失敗が起き,そして組織変性を招き病的状態となる.
3.骨修復と凝固線溶系
1)骨折修復の過程
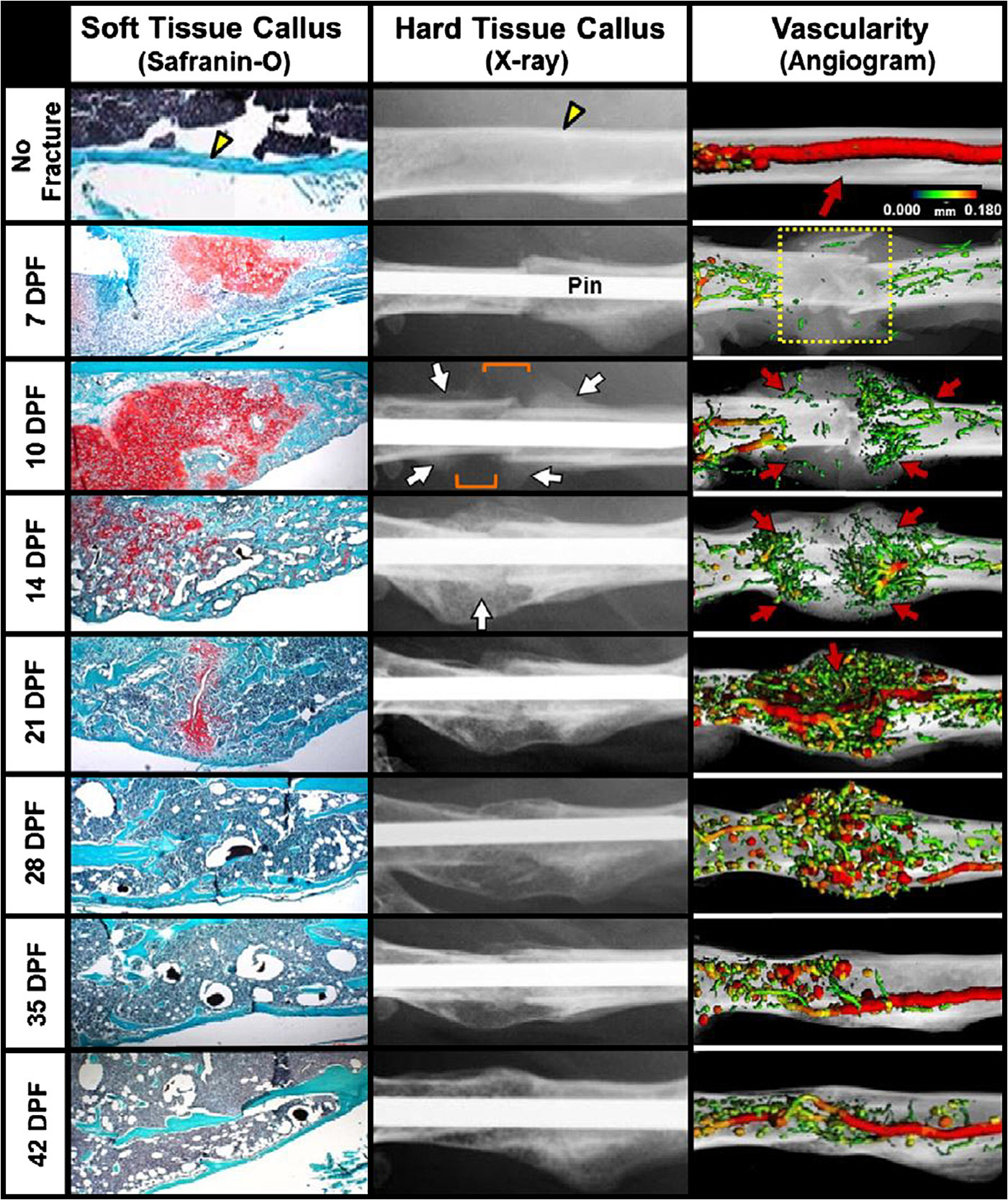
骨化パターンは2通り存在する.膜性骨化と内軟骨性骨化である.膜性骨化は,未分化な細胞から骨芽細胞に分化し,それが骨に変換される骨化形態である.一方,内軟骨骨化は軟骨細胞の形成と増殖・分化後,骨芽細胞によって骨に置換される骨化形態である.両者ともに生体の通常の骨成長と同じプロセスである16, 17).骨の修復,中でも骨折の修復は組織損傷を伴っているという点で骨の成長と異なっている.その修復過程において乗り越えなくてはならない様々な課題が存在する.すなわち,出血,低酸素といった局所の環境の変化である.骨折によって,血行に富んだ骨はまず血管の破綻が発生し,出血が起きる.止血のためにフィブリンが産生される.骨折の治癒の過程は古くから研究されているが18–20),時間的空間的な血管再生の治癒過程を詳細に示したものは少ない.我々はマウスの大腿骨骨折モデルを用いることによって,骨折前は骨内に存在した血行が骨折により骨折部の血行が途絶し,無血管野が形成されることを可視化した16).その無血管野に軟骨細胞が増殖・分化し,軟骨仮骨を形成,その後膜性骨化と内軟骨骨化のプロセスの中で血行が中心部に進展し,骨の連続性が完成すると同時に血行が回復する.骨血行の結合ののち,骨のリモデリングと共に血行もリモデリングしていく.このように血流が骨修復に非常に重要であることが分かる(図1).
2)骨折修復における線溶系の役割
骨折の修復過程の不可欠なプロセスはフィブリンマトリックスの形成といわれている21).骨折が発生すると,損傷部位でのトロンビンがフィブリンと血小板凝集をもたらし出血を止める.そして好中球が治癒促進のため局所の炎症を促進する.フィブリン沈着と好中球活性により,αMβ2インテグリンを表面上にもつマクロファージが活性化し,それがインターロイキン1,6やTNFαといったサイトカイン産生を促す22–24).フィブリンならびに遊走マクロファージの表面に結合したプラスミノゲンはプラスミンに変換される.このプラスミンは組織に蓄積したフィブリンを除去することによって修復過程を促進するとともに抗炎症マクロファージを活性化する23–25).マクロファージによって壊死組織とフィブリン分解産物が除去され,さらに間葉系細胞が浸潤する.さらにプラスミンは骨折部のPro-MMP9やVEGFといった成長因子を活性化し,組織のリモデリングと血管新生を促進する12, 26).すなわち,損傷組織とフィブリンの除去により,一度破断した血管新生が起こり,骨形成が進んでいく.このように線溶系タンパクであるプラスミンはフィブリン依存的メカニズムと非依存的なメカニズムによってこの修復過程に携わっていることが分かる.
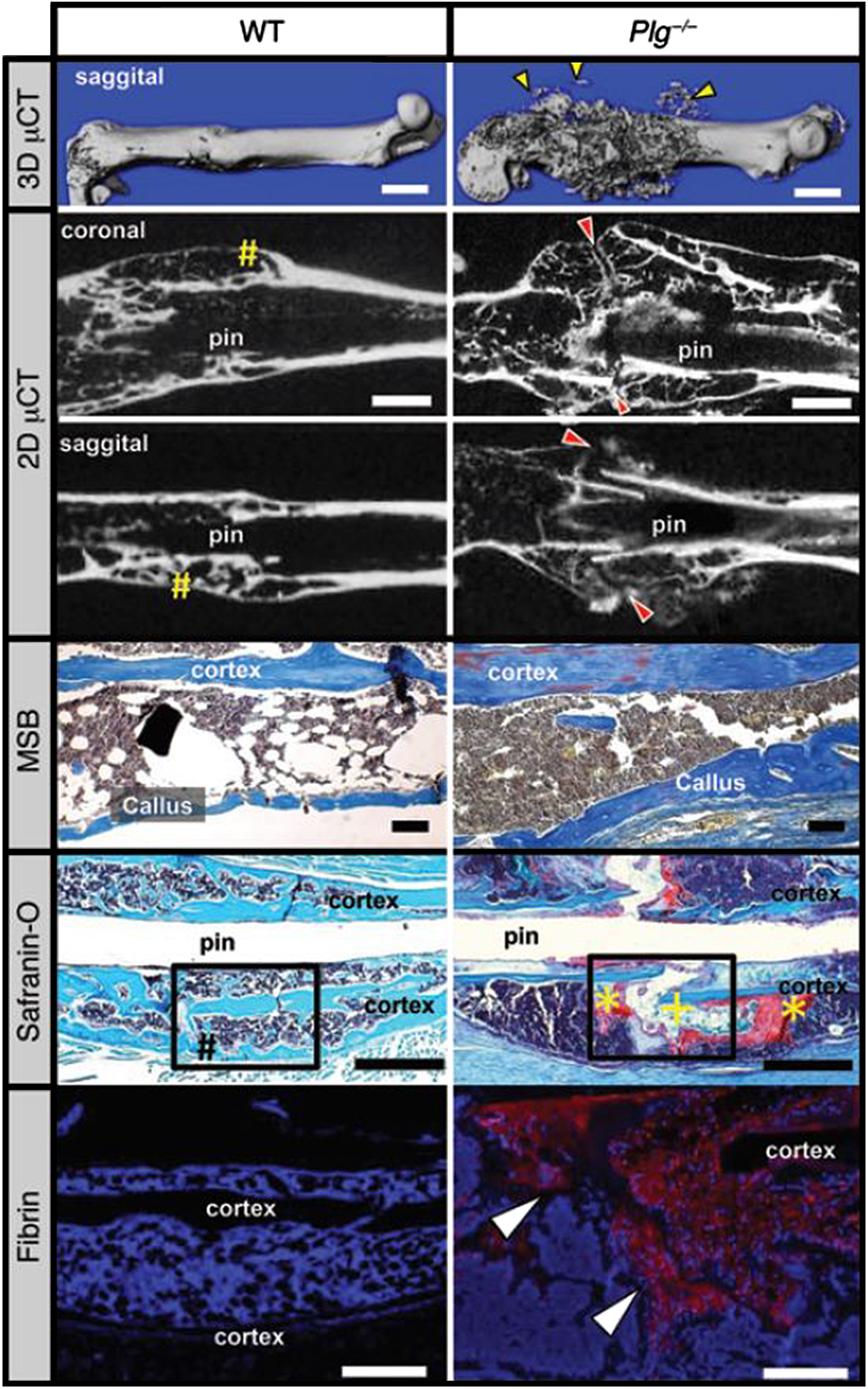
そこで凝固線溶系遺伝子修飾マウスを用いて,骨折修復での凝固と線溶の関与を検証した10).骨折修復初期の不可欠なマトリックスであるといわれるフィブリンの役割を調べるために,フィブリノゲン欠損マウスの骨折修復を検証したところ,骨折後出血はやや多い傾向が見られたが,骨折修復は驚くべきことに正常であった.一方,プラスミノゲン欠損マウスは,骨折修復が損なわれ,偽関節を呈していた(図2).病理組織学的には軟骨形成は両者とも認められるものの,プラスミノゲン欠損マウスにおいて血管の形成が乏しくさらに産生されたフィブリンが除去されずに遺残し,軟骨部と骨形成部との境界にバリアのように存在していた.このプラスミノゲン欠損マウスに対し,薬理学的にフィブリノゲン産生を抑制すると,部分的ではあるが骨折修復が改善した.これらの結果から,骨折修復においてフィブリン分解というフィブリン依存的なプラスミンの作用の他,フィブリン非依存的なプラスミンの作用が存在することが示唆された.
一方,大腿骨骨孔修復モデルにおけるプラスミノゲン欠損マウスにおいては,軟骨形成と骨形成,骨芽細胞増殖が減少していた27).VEGFとTGFβの発現量が低く,血管形成も減少していた.骨修復モデル間で一部相違はあるものの,プラスミノゲンが大いに骨修復に影響があることが示唆された.
線溶系の調節因子であるtPA,uPAやPAI-1の骨折修復寄与の知見も散見される.uPA欠損マウスの骨折は,破骨細胞の移動と血管架橋が乏しく結果として仮骨のリモデリングが乏しかった28).一方tPA欠損マウスにおいて,大腿骨骨欠損モデルにおいて,骨修復が遷延していた29).このメカニズムとして,プラスミン依存的なERK1/2パスウェイが活性化されず骨芽細胞の増殖が抑制されさらに,tPAはHIF1αとVEGFAの活性を高め,新生血管を増加させるということも示された.さらに,PAI-1欠損マウスにおいては,大きなフェノタイプはなく,仮骨の増大とその後速やかな減少が認められるのみであった30).
4.筋修復と凝固線溶系
1)筋修復の過程
人体の筋肉,なかでも骨格筋は関節など骨格を可動させる重要な筋肉である.骨格筋組織は筋線維という多核な巨大細胞から構成される.筋線維の細胞膜と基底膜の間には単核の筋衛星細胞と呼ばれる幹細胞が存在している.この衛星細胞によって骨格筋組織が再生する.骨のようにリモデリングを恒常的に行っているわけではないが,一旦筋組織が損傷すると筋衛星細胞が活性化され,筋芽細胞を経て,筋細胞へと分化する.そして複数の筋細胞が融合し,多核の筋管となり,新たな筋線維が形成され,それがもとの筋線維と融合することによって骨格筋が再生される.
骨損傷と同様,筋肉の損傷後に産生された細胞外基質の溶解が非常に重要で,筋衛星細胞が主な役割を果たす.筋損傷に伴い線維芽細胞や炎症系細胞,マクロファージが損傷組織を貪食する.壊死組織が取り除かれたのち再生血管が起こり,筋衛星細胞が増殖する.炎症系細胞から分泌されるとされるMMP-2やMMP-9といったメタロプロテアーゼが筋再生に寄与しているといわれており8, 31–33),これらMMPsはuPA-プラスミンによるタンパク質分解カスケードによって活性化されるとされている34).
筋肉疾患の病態として,先天的疾患である筋ジストロフィは,筋肉の破壊と変性を繰り返す遺伝的難治疾患であり,最も頻度の高いデュシェンヌ型筋ジストロフィはDMD遺伝子の変異によるジストロフィンの欠損であるとされている.また 後天的疾患としては,筋量減少によるサルコペニア,筋挫傷の修復遅延や筋肉内異所性骨化がある.異所性骨化は,本来形成されるべきでない骨化により,疼痛や可動域の制限などが起き,日常生活を著しく制限する.この病態の原因は諸説あるが35–39),いまだ治療薬開発に至っていない.
2)線溶系と筋肉修復,異所性骨化
プラスミンは筋肉修復と再生にも重要なタンパクであることが示されている34).筋損傷に伴いフィブリンの沈着が起き,その後線溶系機能により溶解される.プラスミン活性は受傷後3~5日でピークに達し,フィブリンと壊死組織を除去し,新たな筋組織を再生する.これはM2マクロファージを通して行われることが分かっている40).沈着したフィブリンなどのデブリスが取り除かれることにより,プラスミンがVEGF-Aやpro-MMPなどといった成長因子,潜在系酵素を活性化し,筋細胞や新生血管を促進する.十分な血行と急性炎症により筋再生が行われる環境が整い,筋管が形成され,筋修復が完成する.デュシェンヌ型筋ジストロフィの動物モデルであるジストロフィン発現を欠損した,DMDマウスは,サルコペニアモデル,筋肉損傷モデルとして,広く筋肉疾患解明に使用されている41).過去の報告ではDMDマウスにおいてMMP-2がVEGFによる血管再生に必要だとする報告31)や,逆にMMP-9を阻害すると筋再生が改善したという報告もある33).
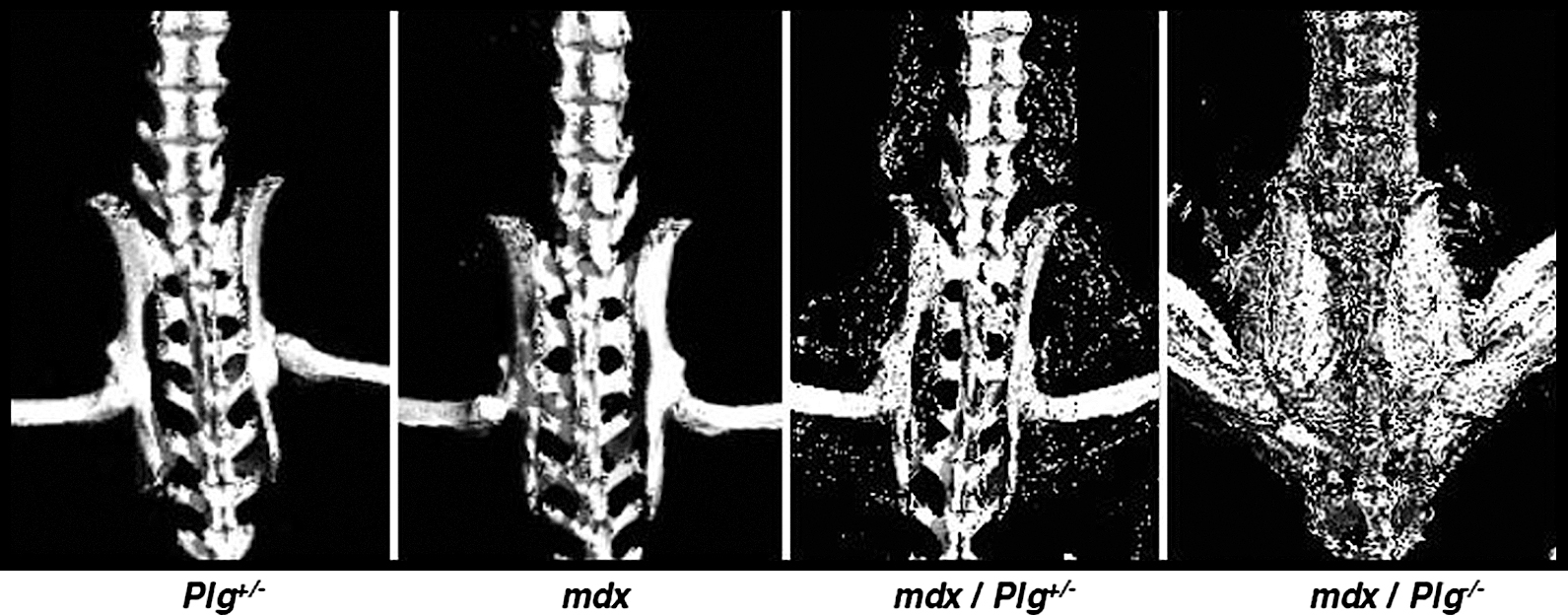
そこで,筋再生促進にDMDマウスの筋損傷におけるプラスミンの意義を調べるべく,プラスミノゲン遺伝子欠損マウスとDMDマウスから,Plg+/– x DMDマウスを作製したところ,驚くべきことに,骨格筋内に石灰化が自然発生した42)(図3).一方,Plg+/–やDMDマウスにおいては,自然発生は起こらなかった.これらの結果から,筋損傷マウスにおける筋修復にプラスミンが関与していることが示唆された.細胞外基質分解酵素が31)プラスミンの作用低下により産生が低下し,損傷部位の筋修復が損なわれ,結果として石灰化を生じたことが示唆された.
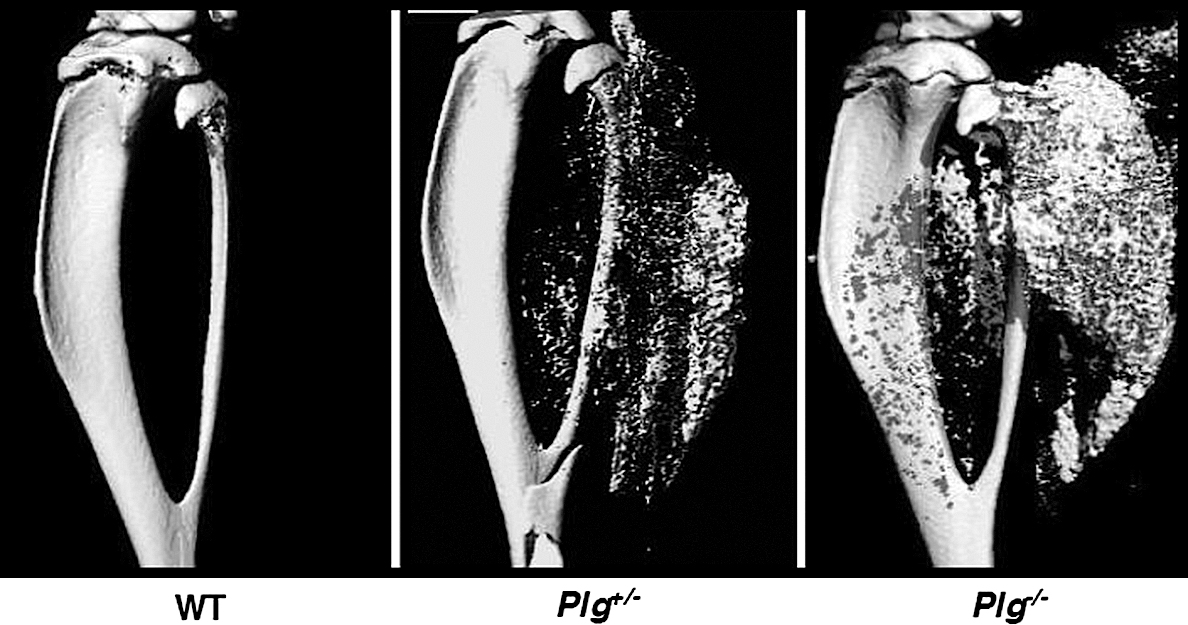
次にプラスミノゲン遺伝子欠損マウスを用いた下腿筋損傷モデルにて検証したところ,プラスミノゲンヘテロマウスとプラスミノゲン欠損マウスにおいて損傷部位の筋肉組織の治癒不良ならびに石灰化をみとめ,一部は成熟骨すなわち異所性骨化が形成された(図4).病理組織学的に検証すると,プラスミンの欠損により,マクロファージの遊走の低下やフィブリンの蓄積による慢性的な炎症がもたらされることが示された.この病態にプラスミン活性が重要であり,約50%のプラスミン活性の低下によって,石灰化をもたらし,異所性骨化を形成するということがわかった.そこでこのマウスに対し,薬理学的にα2プラスミンインヒビターを阻害すると,損傷部の治癒の改善と石灰化を防止できた.さらに,このモデルを用いてフィブリン依存性のプラスミンの役割を検証したところ,この異常石灰化は改善しないことが示された.これら結果から,臨床上において,筋損傷後の正常な筋肉修復と骨化防止に,フィブリン非依存性のプラスミン活性が非常に重要な役割を果たしていることが示唆された.
5.おわりに
線溶系タンパクであるプラスミンの活性は多様な筋骨格系の疾患に大きくに関与していることが示されている.この筋骨格系疾患の今後の展望としては,フィブリン,プラスミンを中心に凝固/線溶システムを修飾し,疾患の治療に結び付けることが肝要である.特に筋骨格系疾患に関してのマクロファージの関与は重要であり,それに働きかけるプラスミン活性のシステムの構築が重要と考えられる.凝固線溶系を修飾することは出血や血栓のリスクも伴う挑戦でもあるが,筋骨格系疾患に対して新たな治療法を模索したい.
著者全員の利益相反(COI)の開示:
本論文発表内容に関連して開示すべき企業等との利益相反なし
文献
- 1) Sundararaghavan V, Mazur MM, Evans B, Liu J, Ebraheim NA: Diabetes and bone health: Latest evidence and clinical implications. Ther Adv Musculoskelet Dis 9: 67–74, 2017.
- 2) Baker CE, Moore-Lotridge SN, Hysong AA, Posey SL, Robinette JP, Blum DM, Benvenuti MA, Cole HA, Egawa S, Okawa A, Saito M, McCarthy JR, Nyman JS, Yuasa M, Schoenecker JG: Bone fracture acute phase response-A unifying theory of fracture repair: Clinical and scientific implications. Clin Rev Bone Miner Metab 16: 142–158, 2018.
- 3) Einhorn TA: Enhancement of fracture-healing. J Bone Joint Surg Am 77: 940–956, 1995.
- 4) Stocum DL: Regenerative biology and medicine. J Musculoskelet Neuronal Interact 2: 270–273, 2002.
- 5) Daci E, Everts V, Torrekens S, Van Herck E, Tigchelaar-Gutterr W, Bouillon R, Carmeliet G: Increased bone formation in mice lacking plasminogen activators. J Bone Miner Res 18: 1167–1176, 2003.
- 6) Daci E, Udagawa N, Martin TJ, Bouillon R, Carmeliet G: The role of the plasminogen system in bone resorption in vitro. J Bone Miner Res 14: 946–952, 1999.
- 7) Enderson BL, Chen JP, Robinson R, Maull KI: Fibrinolysis in multisystem trauma patients. J Trauma 31: 1240–1246, 1991.
- 8) Pepper MS: Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol 21: 1104–1117, 2001.
- 9) Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, Dano K: Impaired wound healing in mice with a disrupted plasminogen gene. Nature Med 2: 287–292, 1996.
- 10) Yuasa M, Mignemi NA, Nyman JS, Duvall CL, Schwartz HS, Okawa A, Yoshii T, Bhattacharjee G, Zhao C, Bible JE, Obremskey WT, Flick MJ, Degen JL, Barnett JV, Cates JMM, Schoenecker JG: Fibrinolysis is essential for fracture repair and prevention of heterotopic ossification. J Clin Invest 125: 3117–3131, 2015.
- 11) Carmo AA, Costa BR, Vago JP, de Oliveira LC, Tavares LP, Nogueira CR, Ribeiro ALC, Garcia CC, Barbosa AS, Brasil BSAF, Dusse LM, Barcelos LS, Bonjardim CA, Teixeira MM, Sousa LP: Plasmin induces in vivo monocyte recruitment through protease-activated receptor-1-, MEK/ERK-, and CCR2-mediated signaling. J Immunol 193: 3654–3663, 2014.
- 12) Roth D, Piekarek M, Paulsson M, Christ H, Bloch W, Krieg T, Davidson JM, Eming SA: Plasmin modulates vascular endothelial growth factor-A-mediated angiogenesis during wound repair. Am J Pathol 168: 670–684, 2006.
- 13) Shen Y, Guo Y, Mikus P, Sulniute R, Wilczynska M, Ny T, Li J: Plasminogen is a key proinflammatory regulator that accelerates the healing of acute and diabetic wounds. Blood 119: 5879–5887, 2012.
- 14) Ploplis VA, French EL, Carmeliet P, Collen D, Plow EF: Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood 91: 2005–2009, 1998.
- 15) Mahmood N, Mihalcioiu C, Rabbani SA: Multifaceted role of the urokinase-type plasminogen activator (uPA) and its receptor (uPAR): Diagnostic, prognostic, and therapeutic applications. Front Oncol 8: 24, 2018.
- 16) Yuasa M, Mignemi NA, Barnett JV, Cates JM, Nyman JS, Okawa A, Yoshii T, Schwartz HS, Stutz CM, Schoenecker JG: The temporal and spatial development of vascularity in a healing displaced fracture. Bone 67: 208–221, 2014.
- 17) Gerstenfeld LC, Cullinane DM, Barnes GL, Graves DT, Einhorn TA: Fracture healing as a post-natal developmental process: Molecular, spatial, and temporal aspects of its regulation. J Cell Biochem 88: 873–884, 2003.
- 18) Rhinelander FW: Tibial blood supply in relation to fracture healing. Clin Orthop Relat Res 1974: 34–81, 1974.
- 19) Rhinelander FW, Baragry R: Microangiography in bone healing. I. Undisplaced closed fractures. The J Bone Joint Surg Am 44-a: 1273–1298, 1962.
- 20) Trueta J: Appraisal of the vascular factor in the healing of fractures of the femoral neck. The Journal of bone and joint surgery British volume 39-b: 3–5, 1957.
- 21) Rockwood CA GD, Bucholz RW: Rockwood and Green’s Fractures In Adults 2010; Philadelphia, Pennsylvania, USA: Wolters Kluwer Health/Lippincott Williams & Wilkins.
- 22) Flick MJ, Du X, Witte DP, Jirousková M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL: Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest 113: 1596–1606, 2004.
- 23) Flick MJ, LaJeunesse CM, Talmage KE, Witte DP, Palumbo JS, Pinkerton MD, Thornton S, Degen JL: Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin alphaMbeta2 binding motif. J Clin Invest 117: 3224–3235, 2007.
- 24) Cole HA, Ohba T, Nyman JS, Hirotaka H, Cates JM, Flick MJ, Degen JL, Schoenecker JG: Fibrin accumulation secondary to loss of plasmin-mediated fibrinolysis drives inflammatory osteoporosis in mice. Arthritis Rheumatol 66: 2222–2233, 2014.
- 25) Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL: Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest 113: 1596–1606, 2004.
- 26) Lijnen HR: Plasmin and matrix metalloproteinases in vascular remodeling. Thrombosis and haemostasis 86: 324–333, 2001.
- 27) Kawao N, Tamura Y, Okumoto K, Yano M, Okada K, Matsuo O, Kaji H: Plasminogen plays a crucial role in bone repair. J Bone Miner Res 28: 1561–1574, 2013.
- 28) Popa NL, Wergedal JE, Lau KH, Mohan S, Rundle CH: Urokinase plasminogen activator gene deficiency inhibits fracture cartilage remodeling. J Bone Miner Metab 32: 124–135, 2014.
- 29) Kawao N, Tamura Y, Okumoto K, Yano M, Okada K, Matsuo O, Kaji H: Tissue-type plasminogen activator deficiency delays bone repair: Roles of osteoblastic proliferation and vascular endothelial growth factor. Am J Physiol Endocrinol Metab 307: E278–288, 2014.
- 30) Rundle CH, Wang X, Wergedal JE, Mohan S, Lau KH: Fracture healing in mice deficient in plasminogen activator inhibitor-1. Calcif Tissue Int 83: 276–284, 2008.
- 31) Miyazaki D, Nakamura A, Fukushima K, Yoshida K, Takeda S, Ikeda S: Matrix metalloproteinase-2 ablation in dystrophin-deficient mdx muscles reduces angiogenesis resulting in impaired growth of regenerated muscle fibers. Hum Mol Genet 20: 1787–1799, 2011.
- 32) Dahiya S, Bhatnagar S, Hindi SM, Jiang C, Paul PK, Kuang S, Kumar A: Elevated levels of active matrix metalloproteinase-9 cause hypertrophy in skeletal muscle of normal and dystrophin-deficient mdx mice. Hum Mol Genet 20: 4345–4359, 2011.
- 33) Li H, Mittal A, Makonchuk DY, Bhatnagar S, Kumar A: Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum Mol Genet 18: 2584–2598, 2009.
- 34) Suelves M, Lopez-Alemany R, Lluis F, Aniorte G, Serrano E, Parra M, Carmeliet P, Muñoz-Cánoves P: Plasmin activity is required for myogenesis in vitro and skeletal muscle regeneration in vivo. Blood 99: 2835–2844, 2002.
- 35) Agarwal S, Loder S, Brownley C, Cholok D, Mangiavini L, Li J, Breuler C, Sung HH, Li S, Ranganathan K, Peterson J, Tompkins R, Herndon D, Xiao W, Jumlongras D, Olsen BR, Davis TA, Mishina Y, Schipani E, Levi B: Inhibition of Hif1α prevents both trauma-induced and genetic heterotopic ossification. Proc Natl Acad Sci U S A 113: E338–347, 2016.
- 36) Forsberg JA, Pepek JM, Wagner S, Wilson K, Flint J, Andersen RC, Tadaki D, Gage FA, Stojadinovic A, Elster EA: Heterotopic ossification in high-energy wartime extremity injuries: Prevalence and risk factors. J Bone Joint Surg Am 91: 1084–1091, 2009.
- 37) Kaplan FS, Le Merrer M, Glaser DL, Pignolo RJ, Goldsby RE, Kitterman JA, Groppe J, Shore EM: Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol 22: 191–205, 2008.
- 38) Nelson ER, Wong VW, Krebsbach PH, Wang SC, Levi B: Heterotopic ossification following burn injury: The role of stem cells. J Burn Care Res 33: 463–470, 2012.
- 39) Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, Hewick RM, Wang EA: Novel regulators of bone formation: Molecular clones and activities. Science 242: 1528–1534, 1988.
- 40) Motley MP, Madsen DH, Jürgensen HJ, Spencer DE, Szabo R, Holmbeck K, Flick MJ, Lawrence DA, Castellino FJ, Weigert R, Bugge TH: A CCR2 macrophage endocytic pathway mediates extravascular fibrin clearance in vivo. Blood 127: 1085–1096, 2016.
- 41) Lu-Nguyen N, Ferry A, Schnell FJ, Hanson GJ, Popplewell L, Dickson G, Malerba A: Functional muscle recovery following dystrophin and myostatin exon splice modulation in aged mdx mice. Hum Mol Genet 28: 3091–3100, 2019.
- 42) Mignemi NA, Yuasa M, Baker CE, Moore SN, Ihejirika RC, Oelsner WK, Wallace CS, Yoshii T, Okawa A, Revenko AS, MacLeod AR, Bhattacharjee G, Barnett JV, Schwartz HS, Degen JL, Flick MJ, Cates JM, Schoenecker JG: Plasmin prevents dystrophic calcification after muscle injury. J Bone Miner Res 32: 294–308, 2017.