Biological nitrogen fixation, the conversion of atmospheric dinitrogen to ammonia, has a significant impact on nitrogen cycles in ecosystems. Nitrogen-fixing microorganisms (diazotrophs) are widely distributed in diverse prokaryotic phyla, but sparsely within these phyla. This distribution pattern suggests that nitrogen fixing ability is evolutionary ancient and mainly transmitted vertically with the widespread loss of function (12). The recent rapid expansion of microbial genome sequences has revealed the presence of the genes encoding homologous proteins to known nitrogenases, even in prokaryotic species that had not previously been recognized as diazotrophs (9). In some cases, experimental evidence for nitrogen fixation was obtained following identification of the responsible genes in the genome (23, 25).
Materials and Methods
Genome survey of nif genes
The genome sequences of bacterial strains in Bacteroidetes were retrieved from the DDBJ database as of July 1st, 2013 (108 genomes) and August 13th, 2014 (an additional 1,238 genomes). The draft genome sequences of 62 strains of Bacteroidetes determined in our laboratories were included in these genomes (for the list of strains and BioProject ID, see http://jcm.brc.riken.jp/en/nbrplist_e). The nif genes were initially searched in these genome sequences with BLAST (1) at a relatively low-level threshold, e-value >10E-20, using nifH and nifD of ‘Ca. A. pseudotrichonymphae’ (locus tags CFPG_545 and CFPG_548, respectively; database accession number AP010656) as queries, and then inspected manually with phylogenetic analyses (see below). This low-level threshold ensured the detection of distantly related sequences even in the outside groups of uncharacterized nif-like sequences. The presence of other nif genes was examined in the selected genome sequences based on the existing annotations and further BLAST searches. The genome sequence of P. propionicigenes WB4T (database accession number CP002345) (11) was included in the analyses as a reference.
Draft genome sequencing
All the bacterial strains used in this study were provided by the Japan Collection of Microorganisms (JCM). These strains were cultured with the conditions specified according to the JCM online catalog database (http://jcm.brc.riken.jp/en/catalogue_e). The draft genome sequences of Bacteroides graminisolvens JCM 15093T and Geofilum rubicundum JCM 15548T were determined, assembled, and annotated with the methods described previously (48).
Acetylene reduction assay
Bacterial strains were cultured with the JCM-specified, nutrientrich medium and the mass of the cultured cells was then inoculated to the nitrogen-poor N2 -fixation medium in a 50-mL stopper bottle. One liter of the N2 -fixation medium comprised 0.688 g of K2 HPO4 , 0.19 g of Na2 SO4 ·10H2 O, 3.75 g of CaCO3 , 30 g of sucrose, a trace amount of biotin, and 1 mL of mineral solution. One liter of the mineral solution comprised 2.4 g of NaMoO4 ·2H2 O, 0.24 g of CoCl2 ·6H2 O, 1.5 g of CaCl2 ·2H2 O, 27 g of FeCl3 ·6H2 O, 28 mL of H2 SO4 , 0.25 g of CuSO4 ·5H2 O, 0.29 g of ZnSO4 ·7H2 O, 1.7 g of MnSO4 ·H2 O, and 12 g of MgSO4 . The gas phase of the medium was initially replaced with N2 and then with 30% of acetylene. After being incubated for 7 d, a 0.1-mL gas sample was assayed after 100-fold dilution with N2 for ethylene production using a gas chromatograph (GC-2014ATC, Shimadzu, Kyoto, Japan) with the Porapak T (80/100 mesh) column (GL Science, Tokyo, Japan) and flame ion detector operating at 50°C and 85°C, respectively. The sensitivity of the measurements was sufficient, even after the dilution. The carrier gas was N2 at a flow rate of 30 mL min−1. The bacterial cell numbers of the cultures were estimated as the most probable numbers.
Phylogenetic analyses
In silico translated amino acid sequences were aligned using MAFFT 7 (22) and manually refined. Only unambiguously aligned residues were used in phylogenetic analyses. Maximum likelihood trees were inferred with RaxML MPI version 8.1.2 (41) using the best model selected with Aminosan in the Kakusan4 package (44). A concatenate sequence analysis was also conducted using the best model for each protein and optimizing the parameter in each protein. Bootstrap analyses of 1,000 replicated re-samplings were conducted to estimate confidence for tree topologies.
Sequence accession numbers
The draft genome sequences of B. graminisolvens JCM 15093T and G. rubicundum JCM 15548T have been deposited in DDBJ/EMBL/GenBank under accession numbers BAJS00000000 and BAZW00000000, respectively.
Results and Discussion
Genome survey of the nif gene
A total of 1,346 genome sequences of Bacteroidetes strains were searched for among homologous genes encoding conventional nitrogenases. In addition to ‘Ca. A. pseudotrichonymphae’ and P. propionicigenes WB4T, homologous genes were detected in the genomes of Dysgonomonas gadei ATCC BAA-286T (ADLV00000000, unpublished), Saccharicrinis fermentans JCM 21142T (BAMD00000000 [43]; recently renamed from Cytophaga fermentans [47]), Bacteroides graminisolvens JCM 15093T, and Geofilum rubicundum JCM 15548T. The homologous genes were also detected in the very recently appeared genomes of Dysgonomonas capnocytophagoides DSM 22835T (NZ_AUFL00000000; unpublished) and Alkaliflexus imshenetskii DSM 15055T (AJUM00000000, unpublished); these two strains were analyzed phylogenetically with their nif genes, but were not assayed for acetylene reduction. The frequency of the nif genes was only 0.5% among the searched genomes. One possible reason for this low frequency is that many genome sequences are still determined with strains associated with humans or animals, and this habitat is likely to be rich in available nitrogen sources.
All these species belong to the order Bacteroidales (class Bacteroidia), and are distributed in three families, Marinilabiliaceae (Saccharicrinis, Geofilum, and Alkaliflexus), Porphyromonadaceae (Dysgonomonas and Paludibacter), and Bacteroidaceae (Bacteroides), among the six recently updated families within Bacteroidales (20). The gene encoding a homologous protein to conventional nitrogenases was not detected in the genome sequences of strains in the other classes in Bacteroidetes (Cytophagia, Flavobacteriia, and Sphingobacteriia), although they accounted for 42.6% of the searched genomes. The isolation sources of these potential diazotrophic species were diverse; human clinical specimens for two Dysgonomonas species (14), rice plant residue in anoxic rice-field soil for P. propionicigenes (45), rice straw residue in a methanogenic reactor for B. graminisolvens (27), deep subsea floor sediment for G. rubicundum (26), marine mud for S. fermentans (4), and an alkaline soda lake for A. imshenetskii (50). They presumably maintained their nitrogen fixation ability for their ecological demands. These isolation sources were not always very poor in available nitrogen; however, if these potential diazotrophs share other habitats poor in nitrogen sources, nitrogen fixation may be of significant importance for their survival and adaptation.
Annotation of draft genomes of B. graminisolvens and G. rubicumdum
The draft genome sequences of B. graminisolvens JCM 15093T and G. rubicumdum JCM 15548T were assembled and annotated in the present study. The total sequence reads of 620,620 for B. graminisolvens and 553,196 for G. rubicumdum were assembled into 63 and 212 contigs with N50 lengths of 128,246 bp and 65,843 bp, 39.0 and 23.4×redundancies, and G+C contents of 41.6% and 44.8%, respectively. The resulting genomes of 3.68 Mbp for B. graminisolvens and 4.92 Mbp for G. rubicumdum contained 3,413 and 4,556 protein coding sequences, respectively.
Identification of functional nif genes
Annotations of the genome sequences revealed the presence of nifH, nifD, nifK, nifE, nifN, and nifB in the eight Bacteroidales genomes. These six genes (nifHDKENB) were clustered in this gene order in the seven genomes, except for A. imshenetskii DSM 15055T (Fig. 1). In A. imshenetskii, nifHDK and nifENB were separated by a distance of 29 kb. These six genes corresponded to the minimum nif gene set proposed as a criterion for predicting nitrogen fixation based on genome sequences (9).
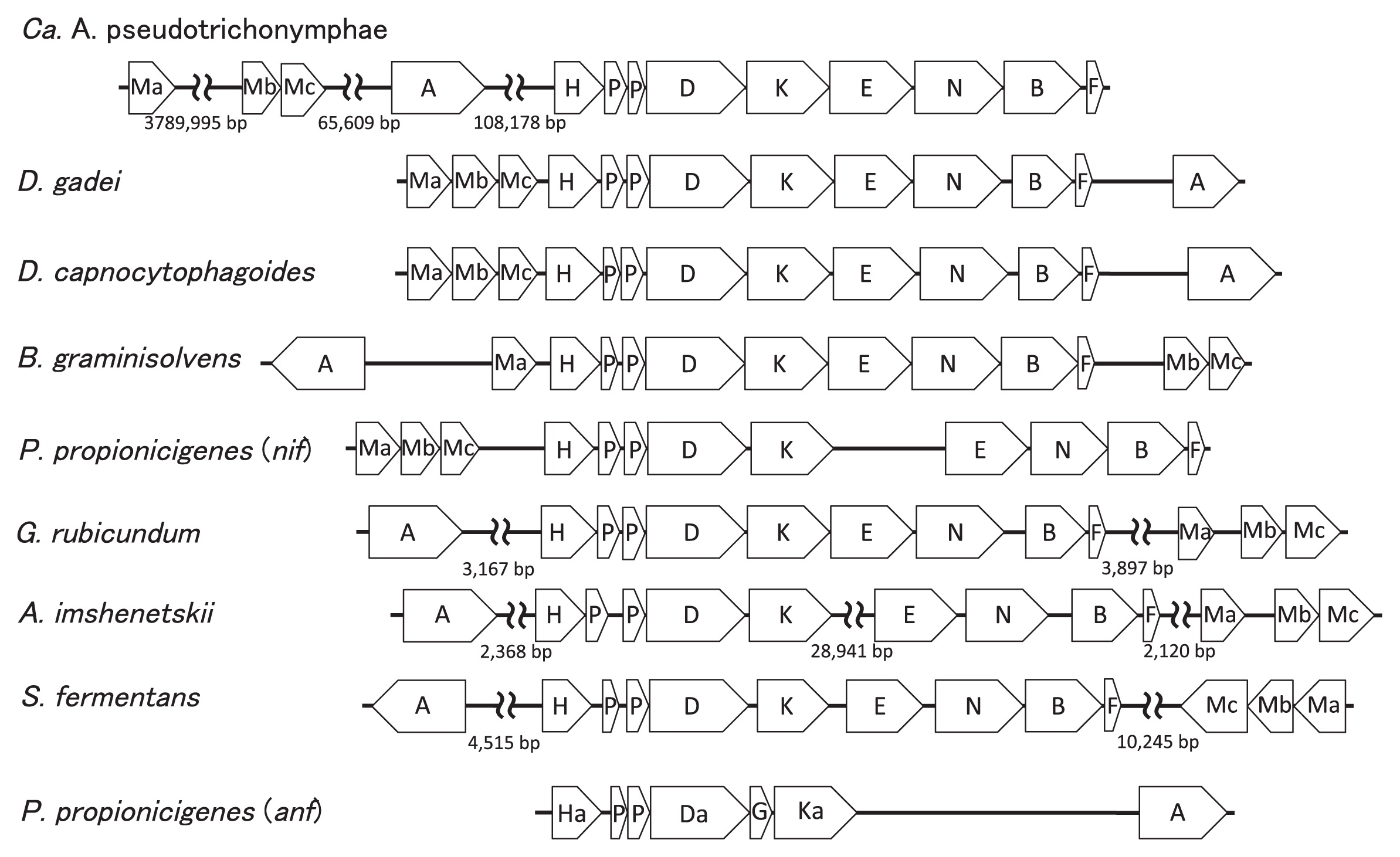
In every genome, the ferredoxin-encoding gene was located downstream of nifB, and two small genes homologous to glnB existed between nifH and nifD, which may be involved in the regulation of nif gene expression (2). The transcriptional regulator gene nifA was sometimes located near the nifHDKENB gene cluster. The genes responsible for molybdenum availability such as modA, modB, and modC were also detected near this gene cluster, but their gene order and relative location to the other genes were not always conserved.
The presence of alternative nitrogenase (anf) genes was previously reported in the genome of P. propionicigenes WB4T (9). In this genome, the anf gene cluster comprised anfHDGK and two glnB homologous genes present between anfH and anfD. The anfHDGK and nifHDKENB clusters were located distantly to each other in the genome (49 kb distance). In our survey of genome sequences, P. propionicigenes WB4T was the only Bacteroidetes member that had the anf gene.
Acetylene reduction activity
Nitrogen fixation ability was examined in five strains that harbored the set of nif genes described above. Acetylene reduction activity was measured for this purpose because this activity was very sensitive and widely used to measure nitrogenase activity. In all of the five examined strains, significant activity, which corresponded to 1/8 to 1/25 that of the diazotrophic strain Clostridium pasteurianum JCM 1408T, was detected after the culture was shifted to the nitrogen poor medium (Table 1). Together with the presence of the nif gene set, these results strongly suggested that these five strains had the ability to fix dinitrogen.
Table 1
Acetylene reduction activity of
Bacteroidales species
| Strain |
Activity (nmol cell−1) |
| Dysgonomonas gadei JCM 16698T |
1.33±0.17×10−6 |
| Bacteroides graminisolvens JCM 15093T |
0.08±0.05×10−6 |
| Paludibacter propionicigenes JCM 13257T |
1.50±0.20×10−6 |
| Geofilum rubicundum JCM 15548T |
0.48±0.04×10−6 |
| Saccharicrinis fermentans JCM 21142T |
0.59±0.04×10−6 |
| Clostridium pasteurianum JCM 1408T |
12.7±0.04×10−6 |
C. pasteurianum JCM 1408T was used as a positive control. Only this strain showed prominent cell growth in the N2 fixation medium. Activity was measured after seven days incubation and expressed as the mean ± standard deviation of three replicated measurements. No activity was detected when Escherichia coli DH5α, Dysgonomonas hofstadii JCM 17038T, and Prevotella paludivivens JCM 13650T were used as negative controls for measurements.
Although evaluating 15N2 stable isotope incorporation is important for providing more direct evidence for nitrogen fixation, the acetylene reduction activities detected were very low and isotope incorporation was not expected. Prominent growth on the nitrogen-poor medium used in this study was not detected for any species, and this may have been because the medium lacked some essential nutrients. The optimization of culturing conditions is necessary in order to further characterize the nitrogen fixation abilities of these species.
Phylogeny of nif gene sequences
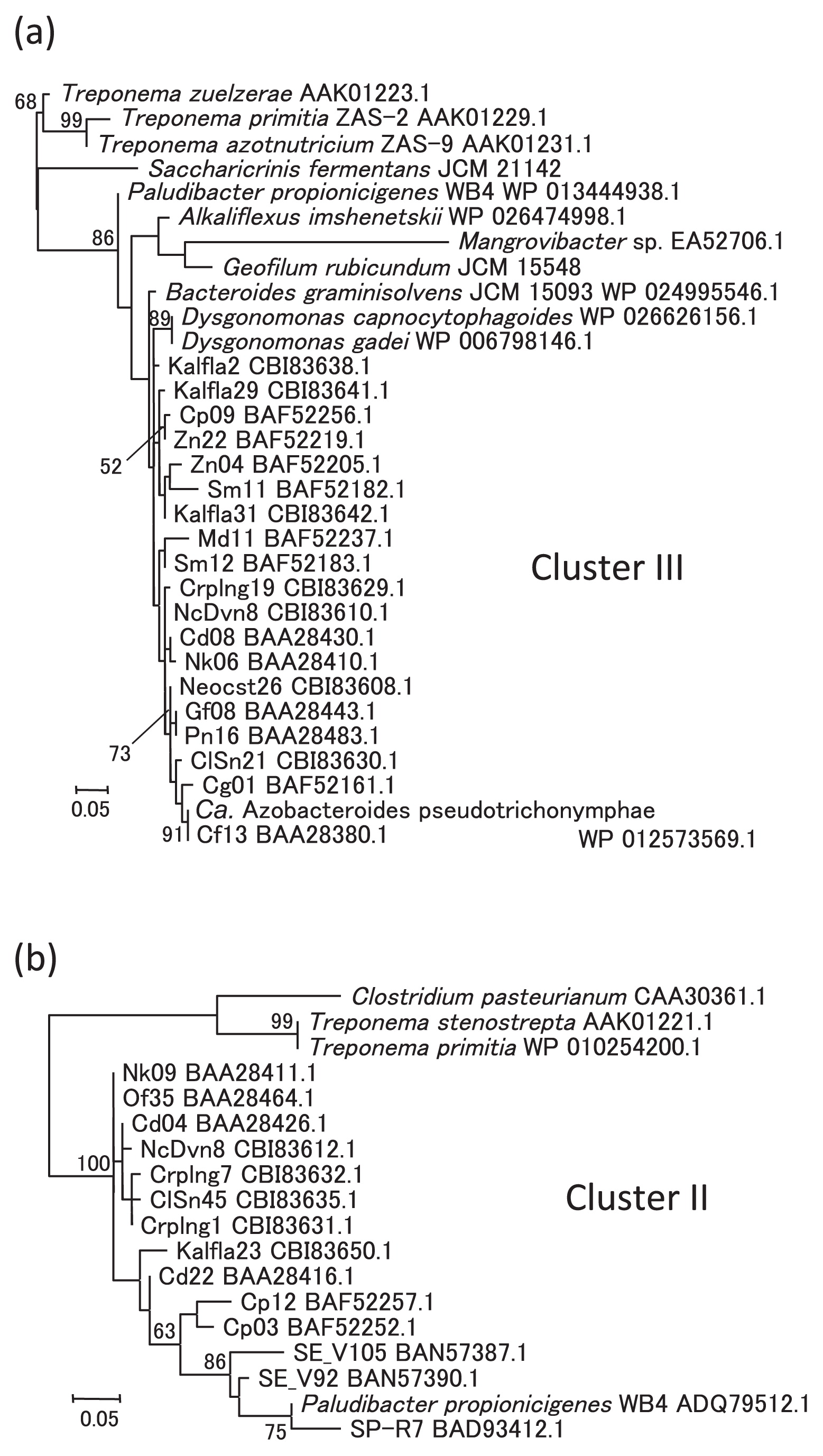
In the phylogenetic tree inferred with the concatenated sequences of the six proteins NifH, NifD, NifK, NifE, NifN, and NifB (Fig. 2), the eight species of Bacteroidales formed a monophyletic cluster in Group III of the nitrogenases defined by Raymond et al. (39) with strong bootstrap support (99%). This result implied that an ancestor of Bacteroidales had the set of nif genes that had been vertically transmitted during their evolution. If this was the case, the sporadic occurrence of nif genes in Bacteroidales members can be attributed to the loss of nif genes in many lineages. The nitrogen fixation reaction requires a large amount of energy, and once species adapted to environments rich in available nitrogen sources, they may have lost the nif genes. The sporadic occurrence of diazotrophs is a common feature among other prokaryotic phyla.
Among the phylogenetic trees of the individual protein sequences (Figs. S1–S6), the monophyly of the eight species occurred in NifK, NifE, and NifB with fairly strong support in the former two (78% and 89%, respectively), but with very weak support in the latter (41%). S. fermentans was located outside the strongly supported monophyletic group of the other seven species in the NifH and NifD trees, whereas S. fermentans branched out very closely and its position was only weakly supported. Three taxa of the genus Treponema were branched within the group of the eight species in the NifN tree; however, these relationships were very weakly supported.
The relationships among the Bacteroidales members were almost fully resolved in the concatenated tree (Fig. 2) and many of these relationships were also observed in the single protein trees (Fig. S1–S6). Except in the NifH tree, ‘Ca. A. pseudotrichonymphae’ and the two Dysgonomonas species were always grouped together with strong support (>99%). ‘Ca. A. pseudotrichonymphae’ and Dysgonomonas species were found to be closely related to each other in an analysis of multiple proteins (M. Yuki, unpublished data), but were distantly related in the 16S rRNA gene sequence analyses reported previously (7, 15, 30, 33). A. imshenetskii and G. rubicundum were sisters to each other in every tree and this relationship was supported fairly in NifH (82%) and strongly in the other trees (100% each). These two species belong to the family Marinilabiliaceae; however, S. fermentans, which belongs to the same family, did not group together in any of the trees. The two Dysgonomonas species and P. propionicigenes, both of which belong to the family Porphyromonadaceae, were also not grouped together, except in the NifK tree. Vertical transmission from a common ancestor may be a general rule for the Bacteroidales nif genes, but not the sole rule for their evolution because strict vertical transmission cannot explain the observed phylogenetic relationship.
Related nif gene sequences in environments
The identification of nif genes in Bacteroidales members facilitates the prediction of organismal origins of related sequences obtained directly from various environments. The nifH sequences were used as queries for searches of related sequences in the DNA sequence database because nifH is often used to detect potential nitrogen fixers in various environments (49). These searches exclusively identified a large number of nifH sequences from the microbial community in the gut of termites as closely related sequences. The sequences from other environments showed less than 90% amino acid identity to the NifH protein sequence of ‘Ca. A. pseudotrichonymphae’.
Termites thrive on nitrogen-poor dead plant materials, and besides cellulose decomposition, nitrogen fixation by the gut bacteria is another crucial aspect of symbiosis with the gut microbial community (6). The diversity of nifH sequences was previously investigated in various termite species and the related cockroach Cryptocercus punctulatus (29, 34, 35, 46), and the relationships between nifH diversity in the gut microbial communities and the lifestyles and phylogenetic positions of host termites have been inferred (46). Although the microorganisms encoding most of these nifH sequences are unknown, the identification of nif genes in Treponema species (phylum Spirochaetes) isolated from a termite gut has enabled us to predict the Treponema origins of some nifH sequences (24). Treponema and Bacteroidales members are major constituents of gut microbial communities and many species, such as ‘Ca. A. pseudotrichonymphae’, are associated with gut cellulolytic protists as endo- or ectosymbionts (17, 37). Bacterial species in Bacteroidales may play a crucial role in symbiotic nitrogen fixation because the nifH and anfH genes related to the Bacteroidales members are some of the most abundant sequences detected in the gut microbial communities of many termite species.
The nifH sequences of Bacteroidales members were closely related to a group of sequences in “Cluster III-1” defined by Yamada et al. (46), and corresponded to the “Bact III-3a” cluster recently defined by Desai and Brune (8), which includes the nifH sequences identified from protist suspensions and predicted to be derived from the ectosymbiont species ‘Candidatus Armantifilum devescovinae’. ‘Ca. A. devescovinae’ and related ectosymbiont species of termite-gut protists, together with the ‘Ca. A. pseudotrichonymphae’ endosymbiont, belong to Cluster V of Bacteroidales, defined based on the 16S rRNA gene sequence (7, 30–33, 36). Therefore, these findings confirmed the ‘Ca. A. devescovinae’ origin of the sequences identified from protist suspensions. The sequences of D. gadei, D. capnocytophagoides, and B. gramnisolvens were also closely related to the sequences from the termite gut, suggesting the presence of related diazotrophic species in addition to species in the Cluster V of Bacteroidales.
The P. propionicigenes anfH sequence was closely related to the sequences represented by the phylotype Nk09 from the termite gut (35, 46) and sequences identified from the protist suspension harboring ‘Ca. A. devescovinae’ (8). These anfH sequences were obtained abundantly from termite species belonging to Kalotermitidae (so-called dry-wood termites), such as Neotermes koshunensis, and the Cryptocercus cockroach. Although Desai and Brune (8) previously reported that this anfH group of sequences may have been due to secondary acquisition in the lineage including ‘Ca. A. devescovinae’, the close relationship with P. propionicigenes anfH strongly suggested vertical transmission of the anf gene from a common ancestor and the loss of anf genes in other lineages such as ‘Ca. A. pseudotrichonymphae’. The preferentially transcribed nature of this group of anf genes in some Kalotermitidae termites (8, 28) can explain the ecological necessity for the retention of the anf genes. P. propionicigenes anfD and anfG also showed high-level amino acid sequence identities to those identified as the preferentially transcribed anf genes in N. koshunensis (82% and 65% identities, respectively). In addition to the sequences from the termite guts, several unpublished nifH sequences from rice roots (e.g. AB184916) and activated sludge (e.g. AB827428) were closely related to P. propionicigenes anfH, suggesting the presence of related species of Bacteroidales as diazotrophs in these environments.
Uncharacterized nif-like genes
In our survey of nif genes in Bacteroidetes genomes, genes encoding proteins slightly homologous, but distantly related to conventional nitrogenases were also found in the genomes of Prevotella bryantii B14T (ADWO00000000) (38) and Bacteroides reticulotermitis JCM 10512T (BAIV00000000) (48). Detailed phylogenetic analyses indicated that their nifD and nifK homologs belonged to so-called nifE-like and nifN-like sequences, respectively, as defined by Dos Santos et al. (9) (see Fig. S7 and S8). These NifE-like and NifN-like proteins are considered to have an as yet unknown function, but not in nitrogen fixation. In these NifE-like and NifN-like sequences, the conserved histidine residue (His 442 in the Azotobacer vinelandii NifD numbering), required for the ligand of iron-molybdenum co-factor co-ordination in conventional nitrogenases (21), was not found despite the presence of the conserved cysteines that coordinate iron-sulfur clusters. Therefore, P. bryantii and B. reticulotermitis were unlikely to have nitrogen fixation abilities.
Furthermore, the nifH homologous gene found in B. reticulotermitis was assigned to the Pseudo-nif group (35) or Group IV (39) of nifH sequences that was also very unlikely to function as nitrogenases. In contrast, P. bryantii had the nifH gene sequence closely related to conventional nitrogenases in Group III and the closest relative was that of Treponema bryantii (data not shown). Both nifE-like and nifN-like sequences of P. bryantii were closely related to those of Fibrobacter succinogenes. Since P. bryantii, T. bryantii, and F. succinogenes are all inhabitants of the rumen of ruminant animals (3, 19, 42), lateral gene transfer may have occurred during their evolution. One of the close relatives of the nifE-like or nifN-like sequences of B. reticulotermitis was that from Clostridium termitidis, and both species were isolated from the gut of termites (13, 40), again implying lateral gene transfer. The clone sequence GFN19 in Pseudo-nif Cluster I from the termite gut (35) and the three clone sequences Cp08, Cp26, and Cp32 from the Cryptocercus cockroach (46) were closely related to the nifH-like sequence of B. reticulotermitis. These clone sequences may have been derived from related species; however, potential lateral gene transfers need to be considered in this prediction.