Abstract
The gastrointestinal tract of mammals is a complex ecosystem with distinct environments and comprises hundreds of different types of bacterial cells. The gut microbiota may play a critical role in the gut health of the host. We herein attempted to identify a microbiota shift that may be affected by porcine epidemic diarrhea (PED). We observed significant differences in microbiota between the control and PED virus (PEDV)-infected groups at both the phylum and genus level. Most commensal bacteria (i.e. Psychrobacter, Prevotella, and Faecalibacterium) in the healthy gastrointestinal tract were decreased due to dysbiosis induced by PEDV infection.
Porcine epidemic diarrhea (PED) is an enteric disease in swine caused by PED virus (PEDV), which is a member of the family Coronaviridae (21). PEDV was first identified in England in 1971 (17), and outbreaks of the virus have since been reported and recently in Europe, Asia, and the USA (4, 20, 25, 27). The first case of a virus outbreak occurred in South Korea in 1992, and has since been followed by yearly outbreaks (11, 24). PED poses the most serious threat to infant piglets, with morbidity and mortality rates previously reported to be between 80 and 100% (19), thereby engendering economics losses in the pork industry.
The gut microbiota is a complex consortium of 10–100 trillion microbial cells in the gastrointestinal tract and influences many aspects of its host (31). Numerous animal studies, including those on mammals, noted the importance of the microbiota for the function and health of the gastrointestinal (GI) tract (8, 18, 29). The GI microbiota at its mature stage is generally considered to be stable with minimal changes in its composition; however, the microbial community is subject to change due to factors including diet, age, or health (13). Due to physiological, nutritional, and immunological contributions of the GI microbiota to the gut health of the host, the structure and functional roles of the biota need to be examined in more detail. We previously reported that some microorganisms were associated with swine health and development (18). Previous studies on PED have mainly focused on sequence-based phylogeny analyses, pathogenicity, treatments, and/or vaccinations, without investigating the relationship between the GI microbiota and PEDV infection. In order to address this critical gap, we herein examined specific microorganisms and/or microbial compositions from fecal samples collected from the large intestine, and compared healthy swine and PEDV infected-swine groups using Illumina MiSeq high-throughput sequencing.
Fourteen fecal samples were collected from two different swine groups: healthy (labeled as normal, n=7) and PEDV infection-diagnosed (designated as infected, n=7) pigs. Pigs were Yorkshire-Landrace-Duroc crossbred pigs from a live-stock farmhouse located in South Korea, and were less than 3 months old at the time of collection. PEDV-infected swine were diagnosed with the infection at the College of Veterinary Medicine at Chungnam National University between 2013 and 2014. All fecal samples were collected from the large intestine and were immediately placed into sterile conical tubes using alcohol-sterilized spatulas, and were then stored at −80°C until later analysis. DNA extraction, a polymerase chain reaction for paired-end sequencing performance, and bioinformatics analysis were described previously (9, 18). Sequencing was performed by the Beijing Genomics Institute (Hong Kong, China) using the MiSeq system (Illumina, San Diego, CA, USA), following the system manufacturer’s instructions. In the bioinformatics analysis, we used modified pipelines as described on the websites: the mothur project (http://www.mothur.org/wiki/MiSeq_SOP) (23) and Quantitative Insights into Microbial Ecology (QIIME) (http://qiime.org/tutorials/illumina_overview_tutorial.html) (3). The bacterial sequence reads were compared with known 16S rRNA genes as reference data obtained from the Ribosomal Database Project (RDP), and were taxonomically assigned based on RDP classifiers (5).
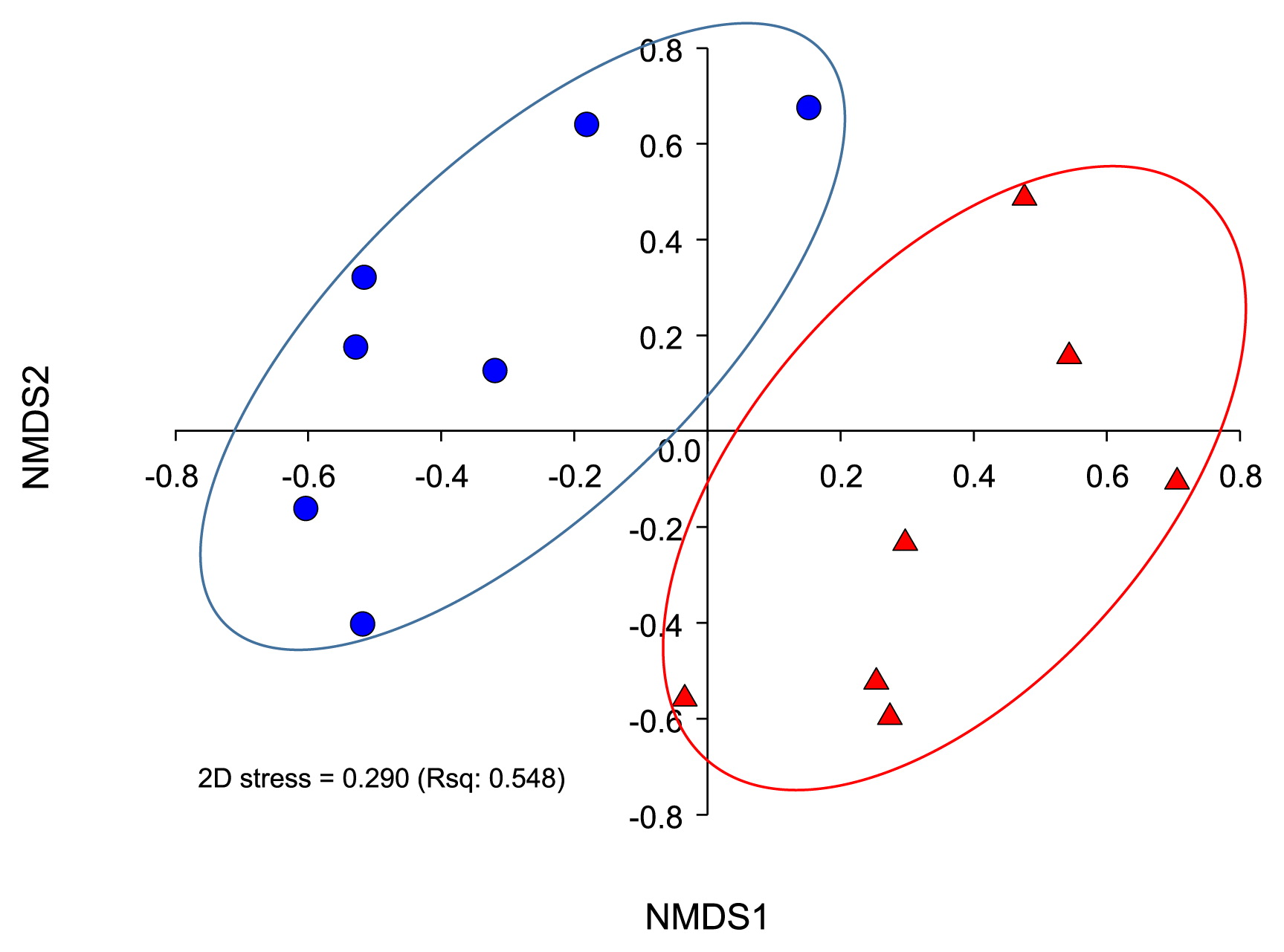
Table 1 summarizes the sequencing reads, diversity indices, and sample coverages of the swine gut samples. After quality control processing and chimeric removal, this study used a total of ca. 600,000 reads (from 646,705 raw reads) to analyze abundance and diversity, as well as to conduct a taxonomic comparison. We used an unweighted pair group method with arithmetic mean clustering (UPGMA) and nonmetric multidimensional scaling (NMDS) analyses, and observed the clear separation of the bacterial community structures of the swine gut into two groups (Fig. 1 and Fig. S1). We also conducted a pairwise comparison analysis of similarity distances of bacterial communities, and the results were consistent with our previous findings obtained using the UPGMA and NMDS method. In the comparison analysis, the gut microbiota of the infected group showed higher similarities than that of the normal group (Fig. S2). According to our measurement of OTUs, Shannon, Chao, and Simpson indices, the gut microbiota diversity of the normal group was higher than that that of the infected group (P<0.05, Table 1). This result was inconsistent with our previous findings, in which the diversity indices for the gut microbiota of a runt swine group and healthy group were higher (18). Using these results, we hypothesized that the diarrhea events of the host caused by PEDV infection may remove or “sweep off” most microorganisms in the gut of the host. PEDV infection-induced dysbiosis may leave host swine afflicted with a range of pathogenic agents.
Table 1
Description of gut microbiota from the large intestinal tract of swine and estimates of sequence diversity and phylotype coverage of MiSeq data. Diversity indices and richness estimators were calculated using the mothur package (of the mother project; http://www.mothur.org). Diversity was estimated using operational taxonomic units (OTUs) and defined as groups with ≥97% sequence similarity. All pigs determined no medical records.
| Sample |
Group |
Gender* |
Total reads |
S & Q** reads |
Analyzed reads |
Observed OTUs |
Shannon |
Chao |
Simpson |
Good’s coverage |
| ped1_16 |
Infected |
F |
97,277 |
45,796 |
44,817 |
438 |
2.249 |
1116.276 |
4.942 |
0.994 |
| ped1_18 |
Infected |
M |
64,239 |
30,262 |
29,743 |
504 |
2.456 |
890.288 |
5.165 |
0.993 |
| ped1_19 |
Infected |
M |
98,613 |
46,187 |
45,435 |
514 |
2.552 |
721.030 |
6.118 |
0.995 |
| ped1_6 |
Infected |
F |
116,735 |
54,949 |
53,016 |
315 |
1.841 |
1713.158 |
3.311 |
0.996 |
| ped1_7 |
Infected |
F |
63,211 |
29,599 |
28,449 |
331 |
2.433 |
988.027 |
6.681 |
0.992 |
| ped1_8 |
Infected |
M |
102,990 |
48,674 |
47,758 |
349 |
2.044 |
954.800 |
4.625 |
0.995 |
| ped1_9 |
Infected |
F |
86,115 |
40,476 |
39,855 |
301 |
2.149 |
844.750 |
3.116 |
0.996 |
| ped2_12 |
Normal |
M |
111,101 |
50,842 |
48,451 |
1106 |
4.585 |
2004.091 |
29.401 |
0.991 |
| ped2_14 |
Normal |
M |
112,492 |
52,925 |
42,505 |
990 |
4.486 |
1957.653 |
34.713 |
0.990 |
| ped2_16 |
Normal |
M |
150,109 |
70,865 |
69,907 |
549 |
0.618 |
1386.545 |
1.187 |
0.995 |
| ped2_18 |
Normal |
F |
120,862 |
56,646 |
43,360 |
937 |
3.579 |
1947.683 |
10.213 |
0.989 |
| ped2_2 |
Normal |
F |
78,684 |
37,220 |
33,552 |
694 |
3.575 |
1614.957 |
13.270 |
0.989 |
| ped2_20 |
Normal |
M |
97,828 |
46,219 |
39,435 |
916 |
4.237 |
1968.683 |
20.823 |
0.989 |
| ped2_8 |
Normal |
F |
76,586 |
36,045 |
33,376 |
730 |
3.964 |
1231.774 |
18.014 |
0.991 |
* F and M denote female and castrated male swine, respectively.
** S & Q denote “subsampled” and “qualified”, respectively.
We observed bacterial communities including typical intestinal bacterial groups in our samples, with the dominant ones being phyla Firmicutes, Bacteroidetes, Proteobacteria, and Fusobacteria, which is consistent with previous findings on swine gut microbiota (8, 10, 14, 18). In the normal group, the phylum Firmicutes marked the highest number of individuals among all microorganisms (approximately 57% of total reads), followed by Proteobacteria (25.1%), Bacteroidetes (10.4%), unclassified Bacteria (3.7%), Actinobacteria (3.2%), and Spirochaetes (0.9%) (Fig. S3). In contrast, the phylum Fusobacteria was dominant in the infected group (approximately 32%, Fig. S3, cf. 0.1% in the normal group). This was interesting because Fusobacteria spp. are typically dominant in the mouth as obligate anaerobic and non-spore-forming bacteria. Fusobacteria spp. have also been frequently identified in a wide variety of clinically significant anaerobic infection cases with other anaerobes, including oral and dental infections (e.g. dental caries), brain abscesses, and tissue infections (2). We gained a more detailed insight by examining results at the phylum and genus levels. At the phylum level, the normal group and PEDV-infected group both had dominance members. However, a difference was detected between the two groups due to a microbial imbalance (i.e. dysbiosis) in the infected group only (28). We also compared genus level taxonomic resolutions between the normal and infected groups. In order to achieve this, we analyzed more than 1% of the total assigned reads and relatively re-calculated the proportions of taxonomic assigned reads (Fig. 2). As expected, a difference was observed in the occurrence and abundance of identified genera between the normal and infected groups. Psychrobacter (45.3%) and Lactobacillus (25.8%) were the most abundant genera in the normal group. Prevotella, Carnobacterium, Faecalibacterium, Sporosarcina, Paenibacillus, and Clostridium genera in that order of abundance were also identified in the normal group. On the other hand, in the infected group, eight genera were more abundant in the gut, including Fusobacterium (30.3%) and Bacteroides (26.9%). With the exception of the genera Psychrobacter and Carnobacterium, the abundances of the genera Fusobacterium, Escherichia (Shigella), Prevotella, and Faecalibacterium significantly differed (P<0.05) between the normal and infected groups. The abundances of pathogenic organisms including Fusobacterium and Escherichia (Shigella) were markedly increased in the infected group (1, 6), while most commensal bacteria (i.e. Psychrobacter, Prevotella, and Faecalibacterium) observed in the healthy gastrointestinal tract decreased. This may have serious implications because these commensal bacteria have been suggested to play important roles such as active digestive enzyme- and/or as short chain fatty acid-producing bacteria. Previous studies indicated that commensal bacteria were crucial for gut health and had functions against diseases including rheumatoid arthritis, ulcerative colitis, and autism (7, 15, 16, 22). One of the commensal bacteria, Psychrobacter spp., as a probiotic, may improve the diversity of the autochthonous gut microbiota by stimulating its growth (26, 30). Furthermore, most species of the genus Carnobacterium function as producers of lactic acid through carbohydrate fermentation and carnobacterium bacteriocins. Moreover, a previous study reported that they had ability to inhibit the growth of Listeria monocytogenes (i.e. food protection) (references therein; 12).

In order to unravel gut microbiota interactions between normal and infected groups, we explored and constructed weighted-OTUs networks. One assumption of this study was that each microorganism (i.e. OTUs) formed a set of consortium within each group, which was proven to be correct by an examination of the profiles of UPGMA, NMDS, and similarity analysis of the gut microbiota (Fig. S4). We observed similarities between the weighted-OTUs network results and other analyses (e.g. NMDS, UPGMA); most OTUs were clearly separated in accord with the group. However, other OTUs were shared within other groups. The latter OTUs are considered common taxa such as Firmicutes and Bacteroides.
This study successfully showed viral infection-induced dysbiosis in the GI tract. Our results substantiate the potential link between the balance of microorganisms in the GI and swine health and development. Future studies are needed in order to determine the functional relationship between diarrhea-causing agents including PEDV and dysbiosis/recovery of the gut microbiota.
The nucleotide sequences obtained in this study were deposited at the EMBL-EBI European Nucleotide Archive under the study accession number PRJEB8875.
Acknowledgements
This work (Grants No. C0250398) was supported by Business for Cooperative R&D between Industry, Academy, and Research Institute funded Korea Small and Medium Business Administration in 2014 and by the Ministry for Food, Agriculture, Forestry and Fisheries, Republic of Korea (Grant no. 112013032SB010).
References
- 1. Afra, K, K Laupland, J Leal, T Lloyd, and D Gregson. 2013. Incidence, risk factors, and outcomes of Fusobacterium species bacteremia. BMC Infect Dis. 13:264.
- 2. Bennett, KW, and A Eley. 1993. Fusobacteria: new taxonomy and related diseases. J Med Microbiol. 39:246-254.
- 3. Caporaso, JG, J Kuczynski, J Stombaugh, et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 7:335-336.
- 4. Chen, JF, DB Sun, CB Wang, HY Shi, XC Cui, SW Liu, HJ Qiu, and L Feng. 2008. Molecular characterization and phylogenetic analysis of membrane protein genes of porcine epidemic diarrhea virus isolates in China. Virus Genes. 36:355-364.
- 5. Cole, JR, B Chai, RJ Farris, Q Wang, AS Kulam-Syed-Mohideen, DM McGarrell, AM Bandela, E Cardenas, GM Garrity, and JM Tiedje. 2007. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 35:D169-D172.
- 6. Croxen, MA, RJ Law, R Scholz, KM Keeney, M Wlodarska, and BB Finlay. 2013. Recent advances in understanding enteric pathogenic Escherichia coli. Clin Microbiol Rev. 26:822-880.
- 7. Kang, DW, JG Park, ZE Ilhan, G Wallstrom, J Labaer, JB Adams, and R Krajmalnik-Brown. 2013. Reduced incidence of Prevotella and other fermenters in intestinal microflora of autistic children. PLoS One. 8:e68322.
- 8. Kim, HB, K Borewicz, BA White, RS Singer, S Sreevatsan, ZJ Tu, and RE Isaacson. 2012. Microbial shifts in the swine distal gut in response to the treatment with antimicrobial growth promoter, tylosin. Proc Natl Acad Sci USA. 109:15485-15490.
- 9. Kim, YS, J Kim, and S-J Park. 2015. High-throughput 16S rRNA gene sequencing reveals alterations of mouse intestinal microbiota after radiotherapy. Anaerobe. 33:1-7.
- 10. Lamendella, R, JW Domingo, S Ghosh, J Martinson, and DB Oerther. 2011. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 11:103.
- 11. Lee, S, and C Lee. 2014. Outbreak-related porcine epidemic diarrhea virus strains similar to US strains, South Korea, 2013. Emerg Infect Dis. 20:1223-1226.
- 12. Leisner, JJ, BG Laursen, H Prevost, D Drider, and P Dalgaard. 2007. Carnobacterium: positive and negative effects in the environment and in foods. FEMS Microbiol Rev. 31:592-613.
- 13. Leser, TD, RH Lindecrona, TK Jensen, BB Jensen, and K Møller. 2000. Changes in bacterial community structure in the colon of pigs fed different experimental diets and after infection with Brachyspira hyodysenteriae. Appl Environ Microbiol. 66:3290-3296.
- 14. Leser, TD, JZ Amenuvor, TK Jensen, RH Lindecrona, M Boye, and K Moller. 2002. Culture-independent analysis of gut bacteria: the pig gastrointestinal tract microbiota revisited. Appl Environ Microbiol. 68:673-690.
- 15. Lopez-Siles, M, TM Khan, SH Duncan, HJ Harmsen, LJ Garcia-Gil, and HJ Flint. 2012. Cultured representatives of two major phylogroups of human colonic Faecalibacterium prausnitzii can utilize pectin, uronic acids, and host-derived substrates for growth. Appl Environ Microbiol. 78:420-428.
- 16. Lucke, K, S Miehlke, E Jacobs, and M Schuppler. 2006. Prevalence of Bacteroides and Prevotella spp. in ulcerative colitis. J Med Microbiol. 55:617-624.
- 17. Oldham, J. 1972. Letter to the editor. Pig Farming October Suppl.:72-73.
- 18. Park, S-J, J Kim, J-S Lee, S-K Rhee, and H Kim. 2014. Characterization of the fecal microbiome in different swine groups by high-throughput sequencing. Anaerobe. 28:157-162.
- 19. Pospischil, A, A Stuedli, and M Kiupel. 2002. Diagnostic notes update on porcine epidemic diarrhea. J Swine Health Prod. 10:81-85.
- 20. Puranaveja, S, P Poolperm, P Lertwatcharasarakul, et al. 2009. Chinese-like strain of porcine epidemic diarrhea virus, Thailand. Emerg Infect Dis. 15:1112-1115.
- 21. Saif, LJ, MB Pensaert, K Sestack, SG Yeo, and K Jung. 2012. Coronaviruses, p.501-524. In JJ Zimmerman, LA Karriker, A Ramirez, KJ Schwartz, and GW Stevenson (ed.), Diseases of Swine, 10th ed. Wiley-Blackwell, Ames, IA.
- 22. Scher, JU, A Sczesnak, RS Longman, et al. 2013. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife. 2:e01202.
- 23. Schloss, PD, SL Westcott, T Ryabin, et al. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 75:7537-7541.
- 24. Song, D, and B Park. 2012. Porcine epidemic diarrhoea virus: a comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes. 44:167-175.
- 25. Stevenson, GW, H Hoang, KJ Schwartz, et al. 2013. Emergence of Porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J Vet Diagn Invest. 25:649-654.
- 26. Sun, YZ, HL Yang, RL Ma, CX Zhang, and WY Lin. 2011. Effect of dietary administration of Psychrobacter sp. on the growth, feed utilization, digestive enzymes and immune responses of grouper Epinephelus coioides. Aquacult Nutr. 17:e733-e740.
- 27. Takahashi, K, K Okada, and K Ohshima. 1983. An outbreak of swine diarrhea of a new-type associated with coronavirus-like particles in Japan. Jpn J Vet Sci. 45:829-832.
- 28. Tamboli, CP, C Neut, P Desreumaux, and JF Colombel. 2004. Dysbiosis in inflammatory bowel disease. Gut. 53:1-4.
- 29. Turnbaugh, PJ, RE Ley, MA Mahowald, V Magrini, ER Mardis, and JI Gordon. 2006. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 444:1027-1031.
- 30. Yang, HL, YZ Sun, RL Ma, JS Li, and KP Huang. 2011. Probiotic Psychrobacter sp. improved the autochthonous microbial diversity along the gastrointestinal tract of grouper Epinephelus coioides. J Aquac Res Development. S1:001.
- 31. Zoetendal, EG, J Raes, B van den Bogert, M Arumugam, CCGM Booijink, FJ Troost, P Bork, M Wels, WM de Vos, and M Kleerebezem. 2012. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 6:1415-1426.
