Abstract
Diarrhea is often associated with marked alterations in the intestinal microbiota, termed dysbiosis; however, limited information is currently available on the intestinal microbiota in captive golden snub-nosed monkeys (Rhinopithecus roxellana) with diarrhea. We herein characterized the fecal microbiota in diarrhea and healthy monkeys using the Illumina MiSeq platform. The concentrations of fecal short-chain fatty acids (SCFAs) and copy numbers of virulence factor genes were also assessed using gas chromatography and quantitative PCR (qPCR), respectively. The results obtained showed that diarrhea monkeys harbored a distinctive microbiota from that of healthy monkeys and had 45% fewer Bacteroidetes. Among healthy subjects, old monkeys had the lowest relative abundance of Bacteroidetes. Linear discriminant analysis coupled with the effect size (LEfSe) and canonical correlation analysis (CCA) identified significant differences in microbial taxa between diarrhea and healthy monkeys. A PICRUSt analysis revealed that several pathogenic genes were enriched in diarrhea monkeys, while glycan metabolism genes were overrepresented in healthy monkeys. A positive correlation was observed between the abundance of nutrition metabolism-related genes and the individual digestive capacities of healthy monkeys. Consequently, the abundance of genes encoding heat stable enterotoxin was significantly higher in diarrhea monkeys than in healthy monkeys (P<0.05). In healthy subjects, adult monkeys had significant higher concentrations of butyrate and total SCFAs than old monkeys (P<0.05). In conclusion, the present study demonstrated that diarrhea had a microbial component and changes in the microbial structure were accompanied by altered systemic metabolic states. These results suggest that pathogens and malabsorption are the two main causes of diarrhea, which are closely related to the microbial structure and functions.
The human/animal gastrointestinal (GI) tract is populated by a complex community of microorganisms that plays a pivotal role in maintaining host health and well-being (10, 49). The intestinal microbiota is crucially involved in extracting nutrients from the diet, which influences host nutrition, metabolism, and body development (50). It may also prevent the colonization of pathogens in the GI tract and is essential for mucosal homeostasis, intestinal maturation, and full functionality (9). Previous studies indicated that a number of GI diseases, including those involving diarrhea, are often associated with specific alterations in microbiota components, called dysbiosis (41). GI diseases are considered to be driven, at least in part, by alterations in the microbiota; however, it currently remains unclear whether dysbiosis is the cause or a consequence of these diseases (11, 33).
The golden snub-nosed monkey (Rhinopithecus roxellana) is a rare and endangered primate under first-class national protection in China. Its distribution is restricted to temperate montane forests at 1,400–3,000 m above sea level across three isolated regions in central and southwestern China (24). Due to their ecological status and research value as a close reference for human health, studies on the health of golden snub-nosed monkeys have attracted worldwide attention. The golden snub-nosed monkey is an Old World monkey in the Colobinae subfamily, which have significantly longer gastrointestinal tracts and larger stomach surface areas than other mammals (20). Wild golden snub-nosed monkeys eat fiber-enriched plants as their staple food (19). However, the diet of captive golden snub-nosed monkeys cannot completely mimic the natural diet of wild moneys, and may contain less fiber or nutrients. Changes in living conditions, particularly in diet, may negatively affect the growth and well-being of golden snub-nosed monkeys, including the induction of gastrointestinal diseases. Gastrointestinal diseases, such as diarrhea, enteritis, and constipation, are the most frequently occurring diseases (60–80%) among captive monkeys, and according to long-term clinical records in Chengdu Zoo, diarrhea is more common in old monkeys. In daily feeding, most captive monkeys have the status of sub-health for long-term or accidental diarrhea, accompanied by decreases in digestion and metabolism, the loss of appetite, and growth retardation. Even though sub-health has not threatened the survival or breeding of golden snub-nosed monkeys, it endangers the health and welfare of this rare species. In order to improve the health of captive golden snub-nosed monkeys, a healthy microbiota in the GI tracts of these monkeys needs to be established in order to prevent diarrhea. Therefore, the present study was conducted in order to characterize the intestinal microbiota of Sichuan golden snub-nosed monkeys in relation their ages and health status. We hypothesize that: i) the components and structures of the intestinal microbiota of healthy and diarrhea monkeys differ, ii) old monkeys have a microbial component that is susceptible to diarrhea, and iii) pathogens and malabsorption are the two main causes of diarrhea and are closely related to the microbial structure and functions.
Materials and Methods
Experimental design and sampling
Our experiment was conducted in the Chengdu Zoo (Chengdu, Sichuan, China), which harbored a captive population of 18 golden snub-nosed monkeys. Their daily diet consisted of approximately 500–800 g of green fodder (privet leaves, mulberry, fruit branches, perennial ryegrass, and Sudangrass), 300–500 g of seasonal fruits and vegetables, and 150–200 g of concentrate (including corn, fish meal, wheat middling, soybean meal, milk, and trace mineral elements). During the experimental period of 18 months, all golden snub-nosed monkeys were monitored with a focus on the occurrence of diarrhea. Sampling work was accomplished by specific zoo keepers in Chengdu for the purpose of animal conservation. All diarrhea samples were collected and its incidence was recorded. Diarrhea was defined as loose or liquid stools, with stools classified as 5, 6, or 7 on the Bristol stool chart (3). Large numbers of samples from healthy animals (from different subjects on the same sampling day or recovered subjects) were also collected. The average life expectancy of golden snub-nosed monkeys in Chengdu Zoo is 12 years and they reach sexual maturity at approximately 3–4 years. Thus, monkeys aged 1–3 years were defined as young monkeys and those aged 4–6 years were adult monkeys. Monkeys older than 6 years and accompanied by decreases in physiological function were defined as old monkeys. Detailed information on sequenced samples was shown in Table S1. Fresh fecal samples were immediately collected upon defecation around feeding time, sealed in sterile plastic bags, transported to the laboratory in liquid nitrogen, and stored at −70°C until analyzed.
Ethics statement
All animals were handled in strict accordance with the animal protection law of the People’s Republic of China (a draft animal protection law was released on September 18, 2009). All procedures were performed in accordance with the rules for the Care and Use of Laboratory Animals of the Animal Ethics Committee of Sichuan Agricultural University (Ya’an, China) (Approval No. 2013-028). All methods were performed in accordance with relevant guidelines and regulations, including any related details.
DNA extraction and sequencing
All samples subjected to DNA extraction were obtained from inside feces using sterile tweezers to avoid soil contamination and had an equal weight of 75 mg. Total genomic DNA was extracted from fecal samples using the E.Z.N.A® Stool DNA Kit (Omega Biotechnology, Norcross, GA, USA) according to manufacturer’s instructions, and stored at −70°C for further analyses. The sequencing of partial 16S RNA genes was performed by Novogene Bioinformatics Technology (Beijing, China). Briefly, DNA was amplified using the 515f/806r primer set (515f: 5′-GTG CCAGCMGCCGCGGTA A-3′, 806r: 5′-XXX XXXGGACTACHV GGGTWT CTA AT-3′), which targeted the V4 region of microbial 16S rRNA, with the reverse primer containing a 6-bp error-correcting barcode unique to each sample. A PCR reaction was performed using the Phusion high-fidelity PCR Master mix (New England Biolabs, Beijing, China) under the following conditions: 94°C for 3 min (1 cycle), 94°C for 45 s/50°C for 60 s/72°C for 90 s (35 cycles), and a last step of 72°C for 10 min. PCR products were purified using the QIAquick Gel Extraction Kit (QIAGEN, Dusseldorf, Germany). Sequencing was conducted on an Illumina MiSeq 2×250 platform according to protocols described by Kozich et al. (25).
Bioinformatics and statistical analysis
Sample reads were assembled by the Mothur software package (44), and then quality filtered and demultiplexed in QIIME using default settings as described by Bokulich et al. (4). Chimeric sequences were removed by UCHIME (14). Operational Taxonomic Units (OTUs) were selected using the de novo OTU picking protocol with a 97% similarity threshold in QIIME software. The taxonomy assignment of OTUs was performed by comparing sequences to the Greengenes database (gg_13_5_otus). A bipartite network was used to display and analyze how OTUs partitioned between samples. Files and statistics of the network were generated by script “make_ otu_network.py” in QIIME. In order to cluster OTUs and samples in the network, a stochastic spring-embedded algorithm was used, as implemented in Cytoscape 3.2.0 (46).
An alpha diversity analysis included the Shannon index, Chao1, and Observed species. Jackknifed beta diversity included unweighted and weighted Unifrac distances calculated with 10 subsampling levels, and these distances were visualized by a Principal Coordinate Analysis (PCoA) (32). The Mann-Whitney U test and type KW test were used as significance tests of alpha diversity. A permutational multivariate analysis of variance (PERMANOVA) and analysis of similarity (ANOSIM) were used as significance tests of beta diversity differences between sample groups. A linear discriminant analysis coupled with the effect size (LEfSe) and canonical correlation analysis (CCA) (5) were performed in order to identify microbial taxa differentially represented between groups at the genus or OTU level (45). The functional profiles of microbial communities were predicted using PICRUSt (27). The bootstrap Mann-Whitney U test and type KW test were also used to identify gene pathways with significantly different abundance between groups. The R packages “Phyloseq”, “biom”, and “pheatmap” were used for data analysis and plotting (37).
Quantitative PCR of virulence factor genes
Virulence factor genes were analyzed using the primers listed in Table S2. qPCR was performed on a Bio-Rad CFX96™ real-time PCR Detection System with CFX Manager Software version 2.0 (Bio-Rad Laboratories, Hercules, CA, USA). Each reaction was run in duplicate in a volume of 25 μL in low 96-well white PCR plates sealed with optical flat 8-cap strips (Bio-Rad Laboratories).
The SybrGreen-based reaction mixture consisted of 12.5 μL SYBR® Premix Ex Taq™ II (TaKaRa Biotechnology, China), 1 μL of each primer (20 mM, Invitrogen Life Technologies, China), and 1 μL of template DNA of fecal samples (the non-template control used water). The amplification program included an initial denaturation at 95°C for 5 min followed by 40 cycles of denaturation at 95°C for 15 s, primer annealing at the optimal temperatures for 30 s and primer extension at 72°C for 30 s, 1 cycle of 95°C for 1 min, 1 cycle at 55°C for 1 min, and a stepwise increase in temperature from 55 to 95°C (at 10 s per 0.5°C) in order to obtain melt curve data. Data were collected at the extension step. Melting curves were checked after amplification and the sizes of PCR products were verified using agarose gels in order to ensure correct amplification results. Standard curves were generated from PCR amplicons according to Metzler-Zebeli et al. (38).
The master mixes of PCR reactions using Taqman probes contained 12.5 μL of Premix Ex Taq (TaKaRa Biotechnology), 2 μL of template DNA, 0.5 μL of each primer, and 1 μL of the probe. The PCR cycle was set to 50°C for 2 min and 95°C for 10 min, followed by 45 cycles at 95°C for 15 s and 60°C for 1 min.
Fecal SCFA analysis
Fecal SCFA concentrations (acetate, propionate, and butyrate) were assessed by gas chromatography as described previously (48).
Results
Metadata and sequencing
Fifty-two fecal samples were collected from diarrhea (n=15) and healthy (n=37) golden snub-nosed monkeys, and heathy subjects consisted of young (n=12), adult (n=12), and old (n=13) monkeys. After removing low quality reads and chimeras, 930,794 high quality reads remained with an average of 17,899.885±7,323.639 tags per sample, ranging between 8,264 and 49,798. These sequences, with an average length of 252 bp, were assigned to 26,182 OTUs based on 97% similarity. In the downstream alpha and beta diversity analyses, the sequence number was normalized to 8,530 by randomly subsampling to standardize sampling efforts. The subsampling of sequences still yielded the sufficient resolution of microbial communities, as suggested by average Good’s coverage, ranging between 91.4±1.1% and 93.2±0.5% (mean±SD).
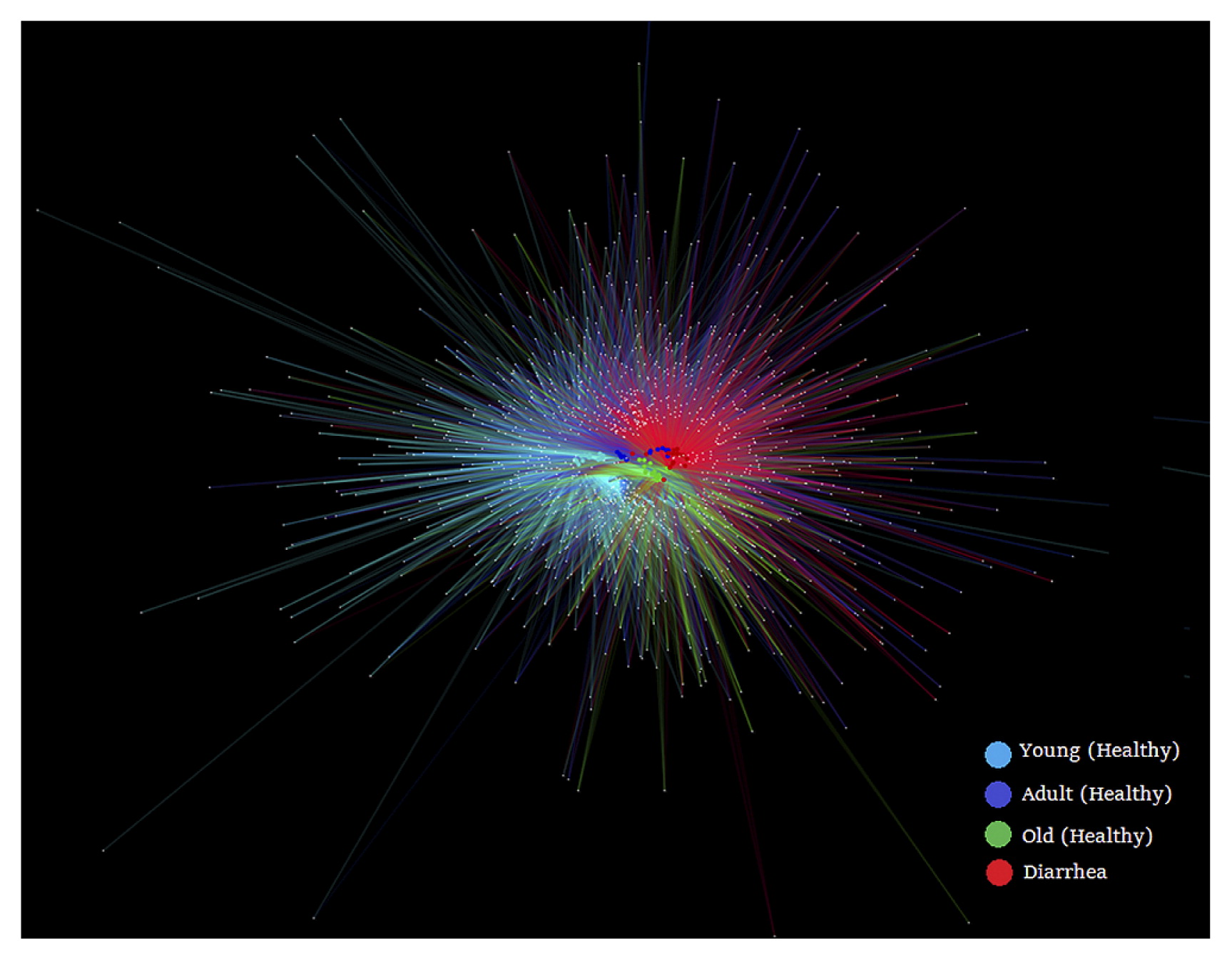
In order to obtain an overview of bacterial genera partitioning across the host, we constructed a bipartite network (Fig. 1). The visual output of this analysis was a clustering of samples according to their shared OTUs, such that samples that share more OTUs clustered closer together. For example, old monkeys (green nodes) were closer to monkeys with diarrhea (red nodes) than other monkeys. This result was consistent with the database of shared OTUs (Fig. S1), which showed that 60.55% OTUs of old monkeys, 57.6% OTUs of adult monkeys, and 50.5% OTUs of young monkeys were shared with monkeys with diarrhea.
Differences in microbial communities between diarrhea and healthy golden snub-nosed monkeys
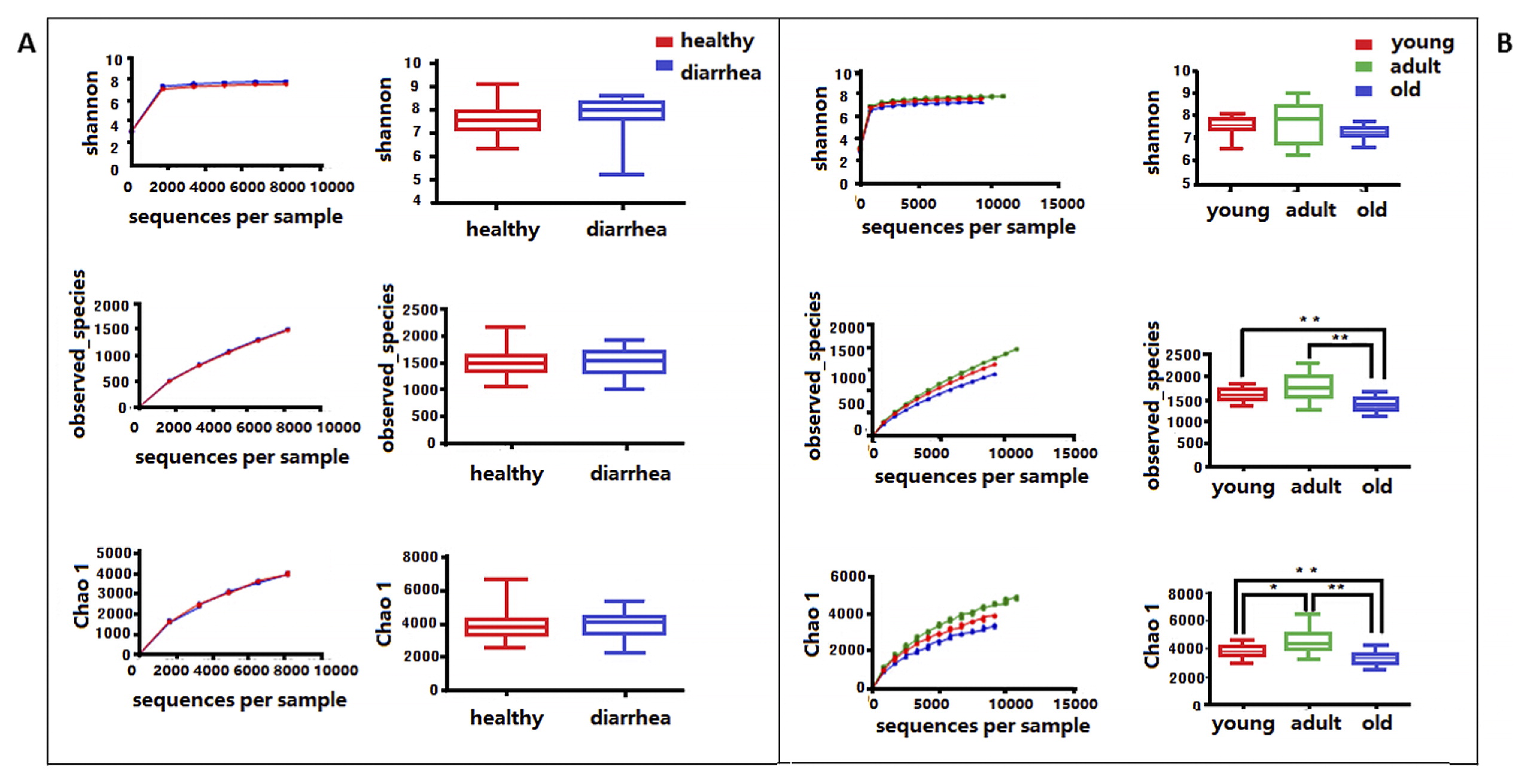
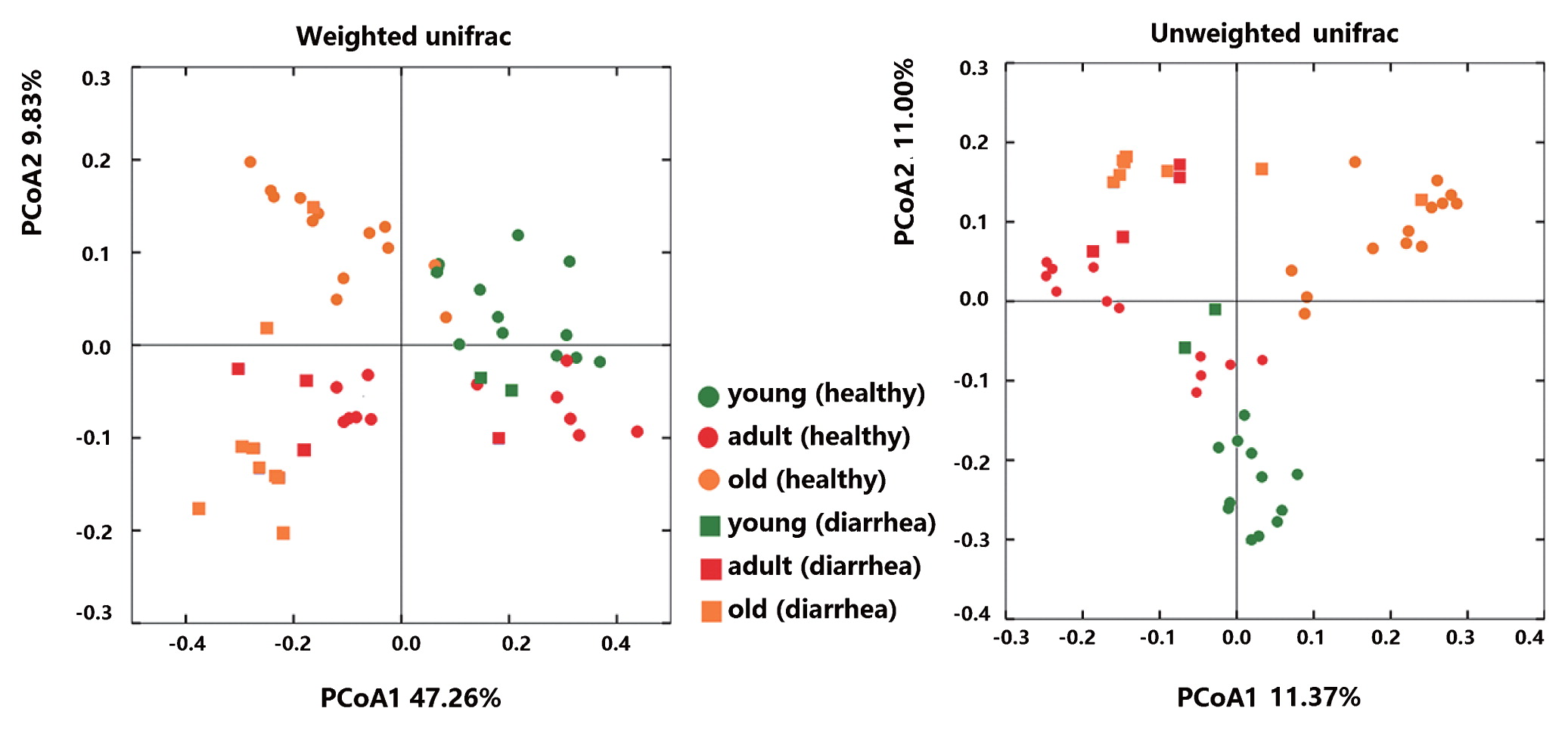
Microbial community richness (alpha diversity) was measured by the Shannon index, Chao1, and Observed species. No significant difference was observed between diarrhea and healthy monkeys (Fig. 2a, bootstrap Mann-Whitney test, P>0.05). In order to examine beta diversity measures between diarrhea and healthy monkeys, weighted and unweighted UniFrac distances were both calculated to estimate dissimilarities in the community membership. PCoA was applied to visualize distances and showed that diarrhea and healthy monkeys harbored distinct microbial taxa (Fig. 3). Based on membership, microbial communities from diarrhea monkeys clustered together and were separate from those from healthy monkeys along the principal coordinate axis. PERMANOVA (F=3.49, P<0.001) revealed significant differences in the community membership between diarrhea and healthy monkeys.
The community compositions of diarrhea and healthy monkeys (genus level) were shown in Fig. 4. Two groups of bacteria were dominant in the intestinal microbiota of captive Sichuan golden snub-nosed monkeys: Bacteroidetes and Firmicutes, which accounted for more than 85% of reads. The relative abundance of these two predominant microbial divisions (phylum level) in monkeys differed between healthy and diarrhea animals: monkeys with diarrhea had 42% fewer Bacteroidetes, and correspondingly more Firmicutes, than those without diarrhea.
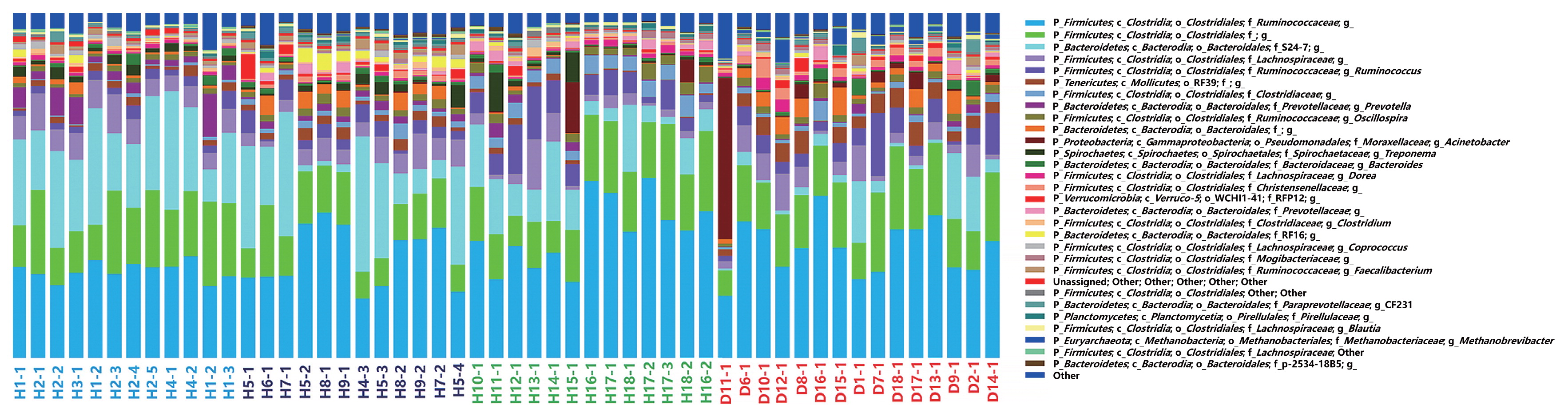
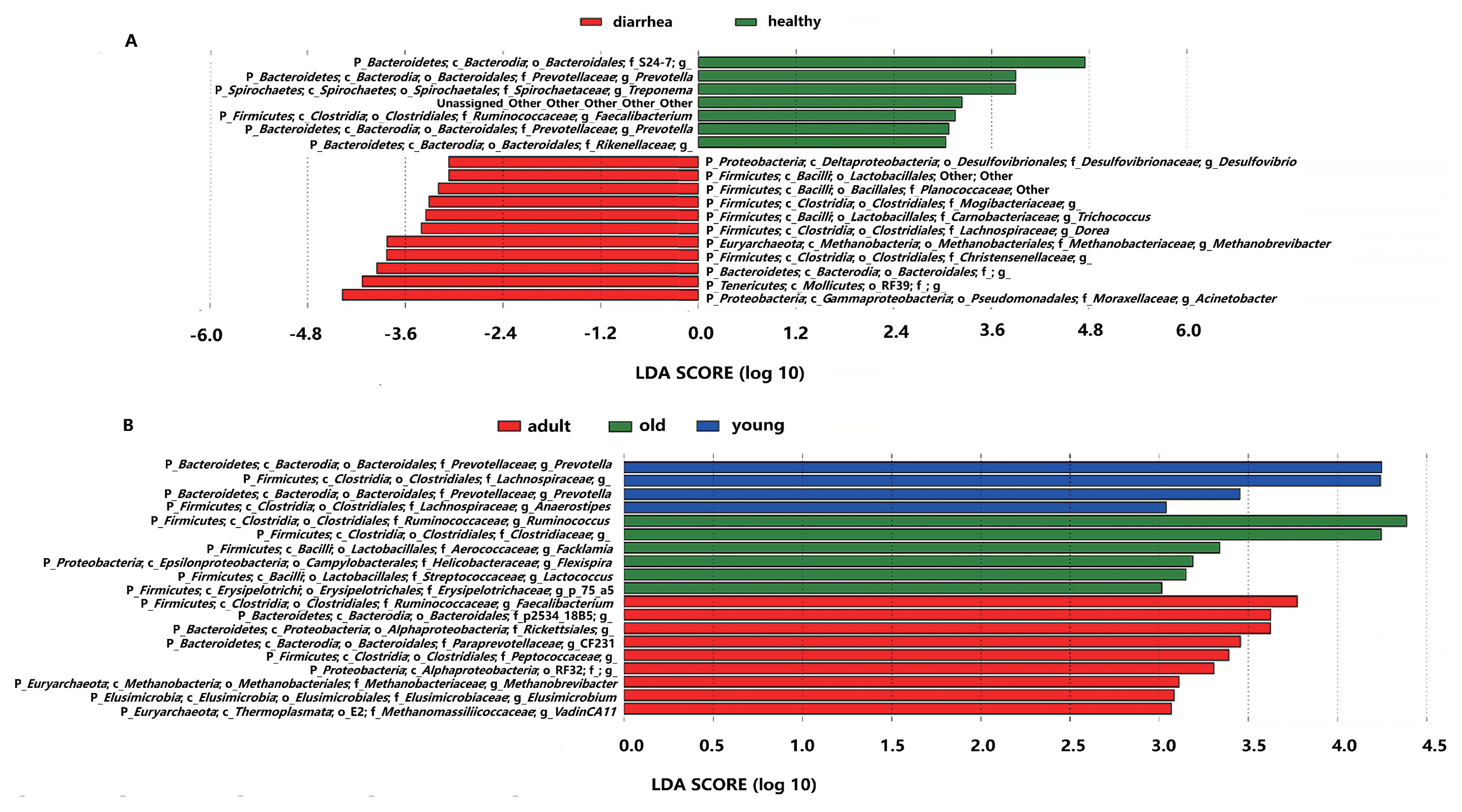
LEfSe was employed to identify specific genera that were differentially distributed between diarrhea and healthy monkeys, and is a robust tool that focuses not only on the significance of differences, but also biological relevance (45). The results of LEfSe were shown in Fig. 5a. Eighteen genera were differentially represented between the two groups, with 11 being more abundant in diarrhea monkeys (e.g., Desulfovibrio spp., Lactobacillales, Planococcaceae, Mogibaceriaceae, Trichococcus spp., Dorea spp., Methanobrevibacter spp., Christensenellaceae, Bacteroidales, RF39, and Acinetobacter spp.) and 7 being more abundant in healthy monkeys (e.g., S24-7., Prevotella spp., Treponema spp., Faecalibacterium spp., and Rikenellaceae). It is important to note that 6 out of 11 genera in diarrhea monkeys were members of the phylum Firmicutes (only 1 out of 7 genera were Firmicutes in healthy monkeys) and 4 out of 7 genera in healthy monkeys belonged to the phylum Bacteroidetes (only 1 out of 11 genera were Bacteroidetes in diarrhea monkeys).
In order to verify the results of the LEfSe analysis, CCA at the OTU level was conducted and the results obtained were shown in Fig. S3. The results of CCA were generally consistent with the LEfSe analysis. For example, the two OTUs (OTU48951 and 33567) close to the factor “diarrhea” belong to the order Clostridiales and genus Acinetobacter, respectively, which were significantly abundant microbial taxa in monkeys with diarrhea in the LEfSe analysis. Similarly, the two OTUs (OTU71071 and 2590) close to the factor “healthy” belonged to the family Ruminococcaceae and S24-7, respectively, which were also significantly abundant microbial taxa in healthy monkeys.
In order to estimate the putative role of intestinal bacteria in the health of monkeys, PICRUSt in combination with QIIME was used to predict the functional composition of a metagenome using marker gene data and a database of reference genomes, which provide direct information on a community’s functional capabilities. Metagenomic inference indicated that diarrhea monkeys harbored microbiomes with a greater abundance of disease-related genes, such as “Staphylococcus aureus infection”, and other genes, including “Metabolism of xenobiotics by cytochrome P450”, “Fluorobenzoate degradation”, “Drug metabolism-cytochrome P450”, “Caprolactam degradation”, “Carotenoid biosynthesis”, and “Retinol metabolism”. Healthy monkeys had more genes such as “Other glycan degradation”, “Glycosphingolipid biosynthesis-ganglio series”, and “Glycosaminoglycan degradation”, as shown in Fig. S4.
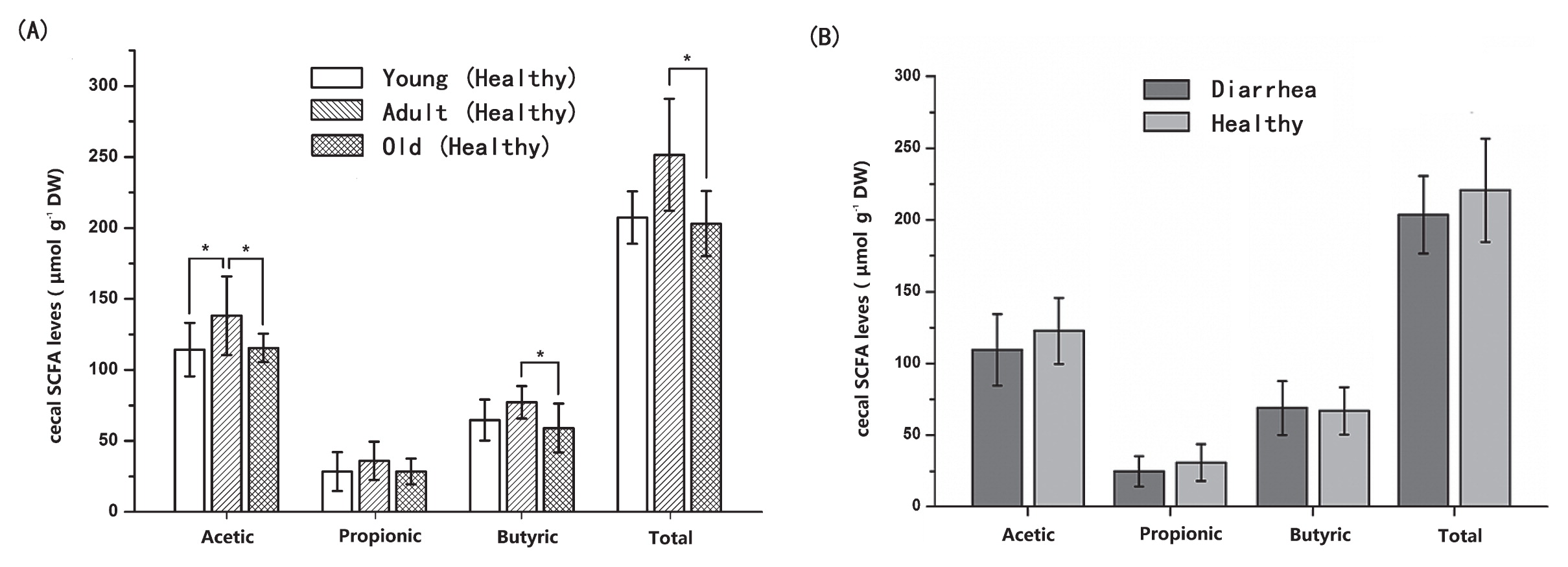
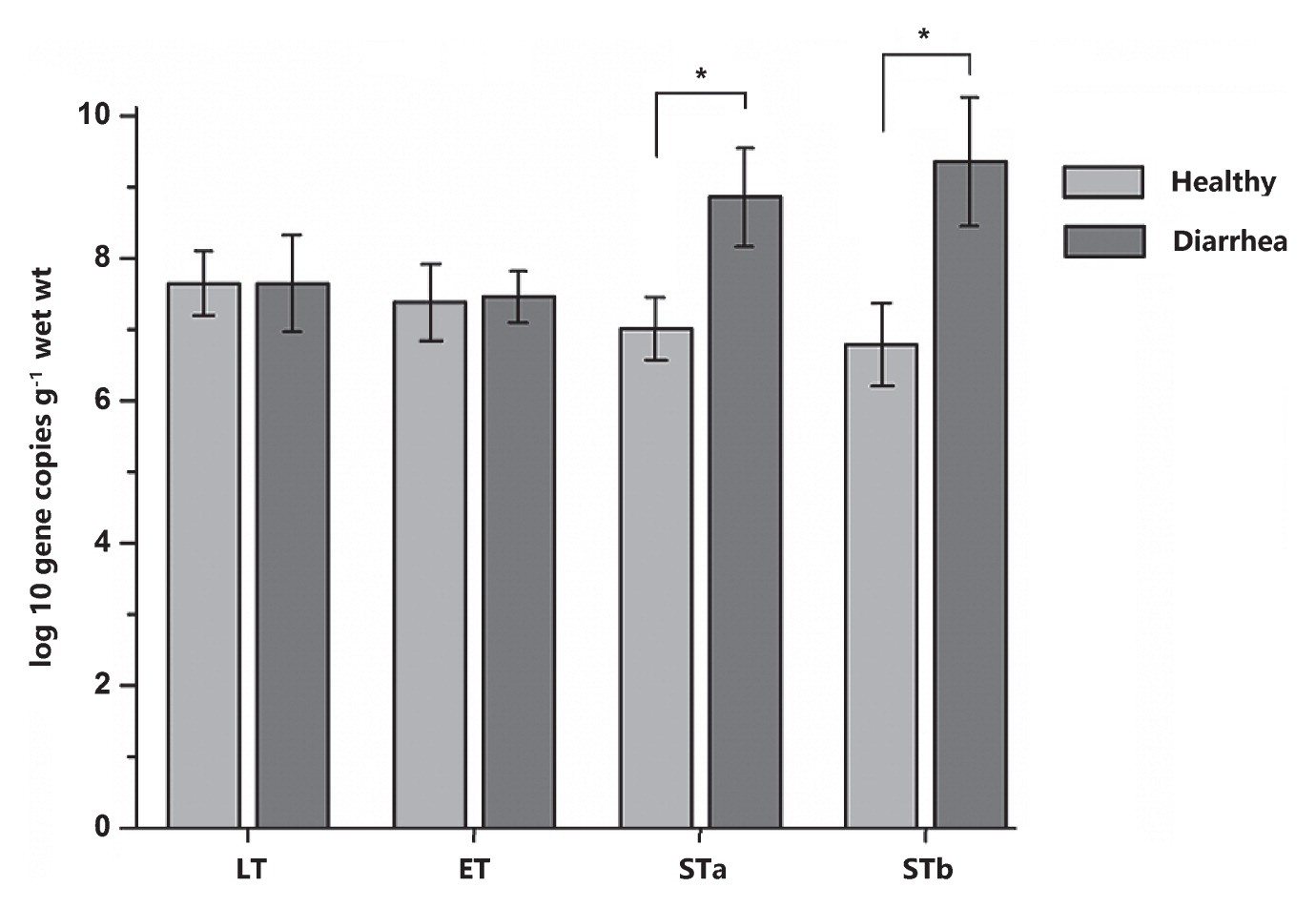
In order to validate the results of PICRUSt, qPCR was employed to detect some common virulence factors genes. The genes encoding the heat-stable enterotoxin of enteroag-gregative E. coli were detected in most samples. Heat labile enterotoxin (LT) and heat stable enterotoxin EAST levels did not significantly differ between diarrhea and healthy monkey, whereas heat stable enterotoxin STa and STb levels were significantly higher in diarrhea monkeys than in healthy monkeys (P<0.05, Fig. 6). Staphylococcal enterotoxin A (SEA) was only identified in two samples from diarrhea monkeys and one sample from healthy monkeys (5.07, 7.10, and 5.57 log10 DNA gene copies g−1 wet weight, respectively). All samples were negative for the Clostridium perfringens alpha-toxin gene (cpA) and staphylococcal enterotoxins D (SED). In the fecal SCFA analysis, no significant differences were observed in the concentrations of acetate, propionate, and butyrate or total SCFAs between diarrhea and healthy monkeys (P>0.05, Fig. 7).
When alpha diversities were compared among healthy golden snub-nosed monkeys of different ages, Chao1 and Observed species indices were significantly lower in old monkeys than in young and adult monkeys (Fig. 2b, type KW test, P<0.01). The bacterial communities of the different age groups were clearly separated from each other by PCoA based on unweighted and weighted Unifrac (Fig. 3). Differences in community memberships among different age groups were also proven to be significant by ANOSIM (r=0.79, P<0.01), indicating distinct microbial community structures among monkeys of different ages. The community compositions of different ages also showed that old monkeys had the lowest relative abundance of Bacteroidetes within healthy subjects (Fig. 4).
The LEfSe analysis was also conducted in order to detect microbial taxa with significantly different abundance among healthy monkeys of different ages (Fig. 5b). Nineteen genera were differentially represented among the three groups (P<0.05), with 4 genera being more abundant in young monkeys (e.g., Prevotella spp., Lachnospiraceae, and Anaerostipes spp.), 9 being more abundant in adult monkeys (e.g., Faecalibacterium spp., P-2534-18B5, Rickettsiales, CF231 spp., Peptococcaceae, RF32, Methanobrevibacter spp., Elusimicrobium spp., and vadinCA11 spp.), and 6 being more abundant in old monkeys (e.g., Ruminococcus spp., Clostridiaceae, Facklamia spp., Flexispira spp., Lactococcus spp., and p-75-a5 spp.). Five out of six genera in old monkeys were members of the phylum Firmicutes. The results of the CCA analysis (Fig. S3) also support the data generated from the LEfSe analysis. For example, OTU29767 was the closest to the factor “old” and belonged to the family Ruminococcaceae, a significantly abundant microbial taxa in old monkeys in the LEfSe analysis.
The PICRUSt analysis was also used to investigate different metabolic potentials among healthy monkeys of different ages. By comparing their predicted metagenomes, the abundance of most nutrition metabolism-related genes appeared in the order “adult>young>old”, which indicated that adult monkeys had a stronger digestive capacity than old monkeys (Fig. S4).
Fecal SCFA concentrations were analyzed as a measure of microbial metabolic activity. Fecal concentrations of acetate were higher in adult monkeys than in young and old monkeys (P<0.05). Although no significant differences were observed in the concentrations of butyrate and total SCFAs between adult and young monkeys (P>0.05), adult monkeys had significant higher concentrations of butyrate and total SCFA than old monkeys (P<0.05, Fig. 7). These results were generally consistent with the results of PICRUSt.
Discussion
In the present study, we characterized the fecal microbiota of Sichuan golden snub-nosed monkeys. The microbiota of golden snub-nosed monkeys is dominated by bacteria belonging to Bacteroidetes and Firmicutes, which account for approximately 90% of all bacteria. At the genus level, the most abundant bacteria also include some known fiber-digesting bacteria, such as Ruminococcus, Prevotella, and Oscillospira (13, 30). This microbiota has enabled these monkeys to adapt to flexible diets including seeds, bamboo buds, fruits, and leaves depending on the availability of food (31).
Diarrhea often causes a significant decrease in microbial diversity in humans and animals (17, 18, 47). When alpha diversities were compared between diarrhea and healthy monkeys, no significant differences were observed in diversity or richness. However, alpha diversity indices were significantly lower in old monkeys than in young and adult monkeys, which implied that the diversity of the intestinal microbiota was relevant to the stability of this micro-ecological system under normal conditions.
Distinct microbial community structures were observed between diarrhea and healthy monkeys. We simultaneously distinguished monkeys of different ages within healthy subjects. Significant differences between diarrhea and healthy subjects, as well as subjects of different ages have been observed in numerous similar studies (2, 22, 34, 39). It is important to note that diarrhea and old subjects shared a similar microbiota in the present study. In the bipartite network and 3D PCoA (Fig. S2), the distribution of nodes representing diarrhea and old subjects were closer. The significance tests of the relative abundance of OTUs, LEfSe and CCA, also revealed that diarrhea and old subjects harbored many common dominant bacteria. In CCA, the factor “old” was close to the factor “diarrhea”.
It was noteworthy that the diarrhea sample in our study was evenly collected from different age groups, although the frequency of diarrhea was the highest in old monkeys. Similarities in microbial communities between old monkeys and diarrhea samples did not appear to be attributed to diarrhea samples being collected more frequently from old monkeys. Sampling work was generally limited by the number of the population and the inhomogeneous age distribution of individuals in the zoo. Moreover, the time of defecation (the acquisition of fresh samples) was random. In the collection of diarrhea samples, multiple sampling was carefully avoided. All of the sequenced samples of diarrhea monkeys came from different individuals. According to our actual execution of the sampling scheme, once a diarrhea sample was collected, fecal samples from other healthy individuals were collected at the same time, as well as after the individual had recovered. This measure was mainly to control variations in the sampling time (seasonal factor) and individual age. Under this condition, multiple sampling was inevitable for the healthy group. However, we considered this sampling method to still be acceptable. Multiple sampling was average and random. More importantly, some of the results of the present study, as well as those of the preliminary experiment revealed by qPCR and DGGE, showed that individual difference factors had less of an influence on the intestinal microbiota than other factors including age, the sampling time, or animal physiology.
As discussed, the most obvious difference between healthy and diarrhea animals was less Bacteroidetes and correspondingly more Firmicutes in diarrhea monkeys than in healthy monkeys. The same changes were observed in old monkeys when they were compared with monkeys of other ages. Increases in the Firmicutes:Bacteroidetes ratio have also recently been reported in humans and animals with diarrhea (18, 47, 52), suggesting that dysbiosis contributed to the occurrence of diarrhea. These results demonstrated that diarrhea had a microbial component, so old monkeys with this microbial structure would be more susceptible to diarrhea.
PICRUSt predicted that the disease-related gene “Staphylococcus aureus infection” was overrepresented in diarrhea monkeys. We previously reported a case of a Sichuan golden snub-nosed monkey infected with Staphylococcus aureus in 2008 (12), but only identified SEA in two samples from diarrhea monkeys. In comparison, enteroaggregative E. coli appeared to be a more serious threat to animal health. The heat-stable enterotoxins of enteroaggregative E. coli were widespread in fecal samples and heat stable enterotoxin STa and STb levels were higher in diarrhea monkeys than in healthy monkeys.
We also found the overrepresentation of glycan metabolism-related genes in healthy monkeys than in diarrhea monkeys (Glycan is one of the most important nutritional components for Sichuan golden snub-nosed monkeys). A positive correlation was observed between the abundance of most nutrition metabolism-related genes and individual digestive capacities (young and adult monkeys had more nutrition metabolism-related genes than old monkeys, and also harbored more powerful digestive capacities). These predictions were validated by the fecal SCFA analysis, in which higher concentrations of SCFAs were observed in adult monkeys. As reported by Hale et al. (20), colobine monkeys, including snub-nosed monkeys, are adapted to leaf eating due to the presence of a forestomach. microbial fermentation in the forestomach plays a crucial role in their higher consumption of mature leaves (higher fiber consumption). Wild golden snub-nosed monkeys consume a number of fiber-enriched plants (large amounts of mature leaves) daily and seasonally, while captive monkeys are confronted by rapid and marked changes in diet (more fruits and concentrates), which significantly alter the gut microbiota and break the micro-ecological balance of forestomach fermentation. Thus, diarrhea in golden snub-nosed monkeys may be associated with a forestomach dysfunction. Our results support the hypothesis that pathogens and malabsorption are the two main causes of diarrhea, which are closely related to the microbial structure and functions.
These results also indicate that Bacteroidetes play a vital role in animal health. Previous studies reported that Bacteroides degrade and ferment organic matter, particularly some soluble or hydrated polysaccharides found in plant cell walls (26, 43). Members of the genus Bacteroides are generally considered to be the most important for pectin and lignin degradation due to their high numbers and nutritional versatility (1, 51). A comparative metagenome survey of the fecal microbiota of a breast- and plant-fed Asian elephant found unexpectedly high diversity in glycoside hydrolase family enzymes and highly abundant polysaccharide utilization loci (PULs) in Bacteroidetes. The baby elephant’s feces sample was dominated by bacteria belonging to Bacteroidetes, which accounted for more than 50% of all bacteria (the S24-7 group of Bacteroidetes accounted for 14%), while the relative abundance of Bacteroidetes (47%) decreased in six-year-old elephants (23).
Previous studies also revealed that species in the genus Bacteroides were involved in the glycosylation of the intestinal epithelium (7), host immune maturation (35), and protective effects against inflammation in animal models of inflammatory bowel disease and multiple sclerosis (36, 40, 42). Moreover, Bacteroides species were shown to be involved in species-specific physical interactions with the host that mediate stable and resilient gut colonization and some mechanisms for symbiosis (28). Decreases in Bacteroides have been widely reported in obesity, colorectal cancer, and enteritis or diarrhea (6, 8, 15–17, 29, 53). Therefore, the actual influence of Bacteroides on the physiological function of the intestines warrants further study, particularly its crucial role in host immunity and the maintenance of intestinal homeostasis.
Lactobacillus and Bifidobacteria are regarded as the most important beneficial bacteria in the GI tract (21). We also observed that the numbers of Lactobacillus and Bifidobacteria were significantly lower in diarrhea monkeys than in healthy monkeys, and were also lower in old monkeys than in monkeys of other ages by qPCR (data not shown). However, when we adopted LEfSe and CCA, due to their low biological relevance, Lactobacillus and Bifidobacterium were not differentially represented in the results of these analyses. Therefore, the relative abundance of Bacteroidetes may reflect the intestinal health status more intuitively, although Bacteroidetes still included some neutral or harmful species. As large taxa, Bacteroides accounted for the most substantial portion of the mammalian gastrointestinal microbiota and played a vital role in the maintenance of intestinal health. Any small change in proportion represented a large alteration in quantity, which had a marked impact on the microbial structure and intestinal ecological functions. Therefore, the relative abundance of Bacteroidetes (e.g., the Firmicutes:Bacteroidetes ratio) has the potential to be an important biomarker for evaluating the health status of the GI.
In conclusion, the results of the present study revealed marked alterations in the intestinal microbiota of golden snub-nosed monkeys with diarrhea. Old monkeys had a similar microbial structure to diarrhea subjects. Fecal dysbiosis was associated with significant changes in the profiles of fecal SCFA and abundance of virulence factor genes, suggesting that pathogens and malabsorption are the two main causes of diarrhea.
Acknowledgements
The present study was supported by the Natural Science Foundation of China (31672318). HZ was supported by the China Scholarship Council.
References
- 1. Akin, D.E. 1988. Biological structure of lignocellulose and its degradation in the rumen. Anim Feed Sci Technol. 21:295-310.
- 2. Ball, J.M., P. Tian, C.Q.Y. Zeng, A.P. Morris, and M.K. Estes. 1996. Age-dependent diarrhea induced by a rotaviral nonstructural glycoprotein. Science. 272:101-104.
- 3. Bishop, S., H. Young, D. Goldsmith, D. Buldock, M. Chin, and R. Bellomo. 2010. Bowel motions in critically ill patients: a pilot observational study. Crit Care Resusc. 12:182-185.
- 4. Bokulich, N.A., S. Subramanian, J.J. Faith, D. Gevers, J.I. Gordon, R. Knight, D.A. Mills, and J.G. Caporaso. 2012. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 10:57-59.
- 5. Bossio, D.A., and K.M. Scow. 1995. Impact of carbon and flooding on the metabolic diversity of microbial communities in soils. Appl Environ Microbiol. 61:4043-4050.
- 6. Broadhurst, M.J., A. Ardeshir, B. Kanwar, J. Mirpuri, U.M. Gundra, J.M. Leung, K.E. Wiens, I. Vujkoviccvijin, C.C. Kim, and F. Yarovinsky. 2012. Therapeutic helminth infection of macaques with idiopathic chronic diarrhea alters the inflammatory signature and mucosal microbiota of the colon. PLoS Pathog. 8:e10030000.
- 7. Bry, L., P.G. Falk, T. Midtvedt, and J.I. Gordon. 1996. A model of host-microbial interactions in an open mammalian ecosystem. Science. 273:1380-1383.
- 8. Chaban, B., M.G. Links, and J.E. Hill. 2012. A molecular enrichment strategy based on cpn60 for detection of epsilon-proteobacteria in the dog fecal microbiome. Microb Ecol. 63:348-357.
- 9. Chow, J., S.M. Lee, Y. Shen, A. Khosravi, and S.K. Mazmanian. 2010. Host-bacterial symbiosis in health and disease. Adv Immunol. 107:243.
- 10. Costello, E.K., C.L. Lauber, M. Hamady, N. Fierer, J.I. Gordon, and R. Knight. 2009. Bacterial community variation in human body habitats across space and time. Science. 326:1694-1697.
- 11. Cremon, C., G. Carini, R. De Giorgio, V. Stanghellini, R. Corinaldesi, and G. Barbara. 2010. Intestinal dysbiosis in irritable bowel syndrome: etiological factor or epiphenomenon? Expert Rev Mol Diagn. 10:389-393.
- 12. Deng, J.B., L.L. Niu, Q. Wang, B. Zhao, X.M. Yu, and W.G. Chen. 2008. Diagnosis and treatment of Golden monkey infected with staphylococcus. Sichuan J Zool. 27:436-437.(In Chinese but abstract in English)..
- 13. Deng, W., D. Xi, H. Mao, and M. Wanapat. 2008. The use of molecular techniques based on ribosomal RNA and DNA for rumen microbial ecosystem studies: a review. Mol Biol Rep. 35:265-274.
- 14. Edgar, R.C., B.J. Haas, J.C. Clemente, C. Quince, and R. Knight. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 27:2194-2200.
- 15. Frank, D.N., A.L.S. Amand, R.A. Feldman, E.C. Boedeker, N. Harpaz, and N.R. Pace. 2007. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 104:13780-13785.
- 16. Goldberg, E., I. Amir, M. Zafran, U. Gophna, Z. Samra, S. Pitlik, and J. Bishara. 2014. The correlation between Clostridium-difficile infection and human gut concentrations of Bacteroidetes phylum and clostridial species. Eur J Clin Microbiol Infect Dis. 33:377-383.
- 17. Gorkiewicz, G., G.G. Thallinger, S. Trajanoski, S. Lackner, G. Stocker, T. Hinterleitner, C. Gully, and C. Hogenauer. 2013. Alterations in the colonic microbiota in response to osmotic diarrhea. PLoS One. 8:e55817.
- 18. Guard, B., J.W. Barr, L. Reddivari, C. Klemashevich, A. Jayaraman, J.M. Steiner, J. Vanamala, and J.S. Suchodolski. 2015. Characterization of microbial dysbiosis and metabolomic changes in dogs with acute diarrhea. PLoS One. 10e0127259.
- 19. Guo, S., B. Li, and K. Watanabe. 2007. Diet and activity budget of Rhinopithecus roxellana in the Qinling Mountains, China. Primates. 48:268-276.
- 20. Hale, V.L., C.L. Tan, K. Niu, Y. Yang, R. Knight, Q. Zhang, D. Cui, and K.R. Amato. 2017. Diet versus phylogeny: a comparison of gut microbiota in captive colobine monkey species. Microb Ecol. 75:1-13.
- 21. Holzapfel, W.H., P. Haberer, J. Snel, U. Schillinger, and J.H. Huis in’t Veld. 1998. Overview of gut flora and probiotics. Int J Food Microbiol. 41:85-101.
- 22. Hopkins, M.J., R. Sharp, and G.T. Macfarlane. 2002. Variation in human intestinal microbiota with age. Dig Liver Dis. 34:S12.
- 23. Ilmberger, N., S. Güllert, J. Dannenberg, U. Rabausch, J. Torres, B. Wemheuer, M. Alawi, A. Poehlein, J. Chow, and D. Turaev. 2014. A comparative metagenome survey of the fecal microbiota of a breastand a plant-fed Asian elephant reveals an unexpectedly high diversity of glycoside hydrolase family enzymes. PLoS One. 9:e106707.
- 24. Kirkpatrick, R., H. Gu, and X. Zhou. 1999. A preliminary report on Sichuan snub-nosed monkeys (Rhinopithecus roxellana) at Baihe Nature Reserve. Folia Primatol. 70:117-120.
- 25. Kozich, J.J., S.L. Westcott, N.T. Baxter, S.K. Highlander, and P.D. Schloss. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 79:5112-5120.
- 26. Kuritza, A.P., P. Shaughnessy, and A.A. Salyers. 1986. Enumeration of polysaccharide-degrading Bacteroides species in human feces by using species-specific DNA probes. Appl Environ Microbiol. 51:385-390.
- 27. Langille, M.G., J. Zaneveld, J.G. Caporaso, D. McDonald, D. Knights, J.A. Reyes, J.C. Clemente, D.E. Burkepile, R.L.V. Thurber, and R. Knight. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 31:814-821.
- 28. Lee, S.M., G.P. Donaldson, Z. Mikulski, S. Boyajian, K. Ley, and S.K. Mazmanian. 2013. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature. 501:426-429.
- 29. Ley, R.E., P.J. Turnbaugh, S. Klein, and J.I. Gordon. 2006. Microbial ecology: Human gut microbes associated with obesity. Nature. 444:1022-1023.
- 30. Ley, R.E., M. Hamady, C.A. Lozupone, P.J. Turnbaugh, R.R. Ramey, J.S. Bircher, M.L. Schlegel, T.A. Tucker, M.D. Schrenzel, and R. Knight. 2008. Evolution of mammals and their gut microbes. Science. 320:1647-1651.
- 31. Ley, R.E., C.A. Lozupone, M. Hamady, R. Knight, and J.I. Gordon. 2008. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol. 6:776-788.
- 32. Lozupone, C., and R. Knight. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 71:8228-8235.
- 33. Lyra, A., T. Rinttila, J. Nikkila, L. Krogiuskurikka, K. Kajander, E. Malinen, J. Matto, L. Makela, and A. Palva. 2009. Diarrhoea-predominant irritable bowel syndrome distinguishable by 16S rRNA gene phylotype quantification. World J Gastroenterol. 15:5936-5945.
- 34. Mariat, D., O. Firmesse, F. Levenez, V.D. Guimarăes, H. Sokol, J. Dore, G. Corthier, and J. Furet. 2009. The firmicutes/bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 9:123.
- 35. Mazmanian, S.K., C.H. Liu, A.O. Tzianabos, and D.L. Kasper. 2005. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 122:107-118.
- 36. Mazmanian, S.K., J.L. Round, and D.L. Kasper. 2008. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 453:620-625.
- 37. McMurdie, P.J., and S. Holmes. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 8:e61217.
- 38. Metzlerzebeli, B.U., S. Hooda, R. Pieper, R.T. Zijlstra, A.G. Van Kessel, R. Mosenthin, and M.G. Ganzle. 2010. Nonstarch polysaccharides modulate bacterial microbiota, pathways for butyrate production, and abundance of pathogenic Escherichia coli in the pig gastrointestinal tract. Appl Environ Microbiol. 76:3692-3701.
- 39. Mueller, S., K. Saunier, C. Hanisch, E. Norin, L. Alm, T. Midtvedt, A. Cresci, S. Silvi, C. Orpianesi, and M.C. Verdenelli. 2006. Differences in fecal microbiota in different european study populations in relation to age, gender, and country: a cross-sectional study. Appl Environ Microbiol. 72:1027-1033.
- 40. Ochoareparaz, J., D.W. Mielcarz, L.E. Ditrio, A.R. Burroughs, S. Begumhaque, S. Dasgupta, D.L. Kasper, and L.H. Kasper. 2010. Central nervous system demyelinating disease protection by the human commensal bacteroides fragilis depends on polysaccharide A expression. J Immunol. 185:4101-4108.
- 41. Packey, C.D., and R.B. Sartor. 2009. Commensal bacteria, traditional and opportunistic pathogens, dysbiosis and bacterial killing in inflammatory bowel diseases. Curr Opin Infect Dis. 22:292.
- 42. Round, J.L., and S.K. Mazmanian. 2010. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci USA. 107:12204-12209.
- 43. Salyers, A.A., J.R. Vercellotti, S.E. West, and T.D. Wilkins. 1977. Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl Environ Microbiol. 33:319-322.
- 44. Schloss, P.D., S.L. Westcott, T. Ryabin, J.R. Hall, M. Hartmann, E.B. Hollister, R.A. Lesniewski, B.B. Oakley, D.H. Parks, and C.J. Robinson. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 75:7537-7541.
- 45. Segata, N., J. Izard, L. Waldron, D. Gevers, L. Miropolsky, W.S. Garrett, and C. Huttenhower. 2011. Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60.
- 46. Shannon, P., A. Markiel, O. Ozier, N.S. Baliga, J.T. Wang, D. Ramage, N. Amin, B. Schwikowski, and T. Ideker. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13:2498-2504.
- 47. Suchodolski, J.S., M.L. Foster, M. Sohail, C.M. Leutenegger, E.V. Queen, J.M. Steiner, and S.L. Marks. 2015. The fecal microbiome in cats with diarrhea. PLoS One. 10:e0127378.
- 48. Sugawara, T., and T. Miyazawa. 2001. Beneficial effect of dietary wheat glycolipids on cecum short-chain fatty acid and secondary bile acid profiles in mice. J Nutr Sci Vitaminol. 47:299-305.
- 49. Turnbaugh, P.J., R.E. Ley, M. Hamady, C.M. Fraser-Liggett, R. Knight, and J.I. Gordon. 2007. The human microbiome project. Nature. 449:804-810.
- 50. Turnbaugh, P.J., and J.I. Gordon. 2009. The core gut microbiome, energy balance and obesity. J Physiol (Oxford, U K). 587:4153-4158.
- 51. Weaver, J., T.R. Whitehead, M.A. Cotta, P.C. Valentine, and A.A. Salyers. 1992. Genetic analysis of a locus on the Bacteroides ovatus chromosome which contains xylan utilization genes. Appl Environ Microbiol. 58:2764-2770.
- 52. Youmans, B.P., N.J. Ajami, Z.D. Jiang, F. Campbell, W.D. Wadsworth, J.F. Petrosino, H.L. Dupont, and S.K. Highlander. 2015. Characterization of the human gut microbiome during travelers’ diarrhea. Gut Microbes. 6:110-119.
- 53. Zackular, J.P., M.A.M. Rogers, M.T. Ruffin, and P.D. Schloss. 2014. The Human gut microbiome as a screening tool for colorectal cancer. Cancer Prev Res. 7:1112-1121.