Abstract
Chitin amendment is an agricultural management strategy for controlling soil-borne plant disease. We previously reported an exponential decrease in chitin added to incubated upland soil. We herein investigated the transition of the bacterial community structure in chitin-degrading soil samples over time and the characteristics of chitinolytic bacteria in order to elucidate changes in the chitinolytic bacterial community structure during chitin degradation. The addition of chitin to soil immediately increased the population of bacteria in the genus Streptomyces, which is the main decomposer of chitin in soil environments. Lysobacter, Pseudoxanthomonas, Cellulosimicrobium, Streptosporangium, and Nonomuraea populations increased over time with decreases in that of Streptomyces. We isolated 104 strains of chitinolytic bacteria, among which six strains were classified as Lysobacter, from chitin-treated soils. These results suggested the involvement of Lysobacter as well as Streptomyces as chitin decomposers in the degradation of chitin added to soil. Lysobacter isolates required yeast extract or casamino acid for significant growth on minimal agar medium supplemented with glucose. Further nutritional analyses demonstrated that the six chitinolytic Lysobacter isolates required methionine (Met) to grow, but not cysteine or homocysteine, indicating Met auxotrophy. Met auxotrophy was also observed in two of the five type strains of Lysobacter spp. tested, and these Met auxotrophs used d-Met as well as l-Met. The addition of Met to incubated upland soil increased the population of Lysobacter. Met may be a factor increasing the population of Lysobacter in chitin-treated upland soil.
Chitin, which is a polymer of N-acetylglucosamine, is the most abundant renewable natural resource after cellulose (Muzzarelli, 1977; Deshpande, 1986; Gooday, 1991). It is widely distributed in nature, particularly in marine invertebrates, insects, fungi, and algae (Nicol, 1991). The supplementation of soil with chitin effectively suppresses plant-pathogenic fungi (Mitchell and Alexander, 1962; Buxton et al., 1965; Khalifa, 1965; Henis et al., 1967; Sneh and Henis, 1970; Van Eck, 1978; Murakami et al., 2009; Cretoiu et al., 2013) and nematodes (Mian et al., 1982; Sarathchandra et al., 1996). In most, but not all cases, the mechanisms underlying this suppressive activity often involve a change in the structure and/or activity of the microbiota in the soil, which subsequently suppresses plant pathogens (Weller et al., 2002; Mendes et al., 2011). Chitinolytic microorganisms that hydrolyze the chitinous hyphae of pathogenic fungi increase their numbers and activity in response to the chitin added. Alternatively, secondary responders to added chitin may suppress pathogens (Cretoiu et al., 2013).
Previous studies that isolated microbial community members from soils showed that Streptomyces, Micromonospora, Nocardia of Actinomycetes, Achromobacter, Flavobacterium,
Chromobacterium, Bacillus, Cytophaga, and Pseudomonas were representative genera of chitinolytic bacteria, while Aspergillus and Mortierella were isolated as chitinolytic fungi (Veldkamp, 1955). 16S rRNA gene analyses revealed that Actinobacteria and Oxalobacteraceae populations and their ability to suppress plant pathogens increased in chitin-treated soils (Cretoiu et al., 2013). Another study demonstrated that chitin enrichment led to an increase in Actinobacteria, γ-Proteobacteria, and β-Proteobacteria, suggesting the specific selection of these chitin-degrading bacteria (Jacquiod et al., 2013). Various chiA genotypes including β-Proteobacteria, γ-Proteobacteria, Actinobacteria,
Acidobacteria, Bacteroidetes, Firmicutes, Planctomycetes, and Chloroflexi were shown to respond to chitin supplementation in soil slurries (Wieczoreck et al., 2014). However, limited information is currently available on temporal changes in the bacterial community structure of chitin-supplemented soil.
We previously investigated the degradation profile of chitin in incubated upland soil and found that added chitin exponentially decreased over time (Kumeta et al., 2018). We herein aimed to identify the chitinolytic bacterial genera involved in exponential chitin degradation using the same soil samples. We performed an amplicon sequencing analysis to investigate the mechanisms by which the bacterial community structure is altered during exponential chitin degradation. We also characterized the chitinolytic bacterial strains isolated from soil samples.
Materials and Methods
Preparation of soil samples
We used soil samples prepared for our previous study that was conducted to investigate chitin degradation profiles (Kumeta et al., 2018). The surface soil of Brown Forest soil, classified as light clay, in an upland field was air-dried and passed through a sieve with 2-mm meshes. Chitin powder (Wako Chemicals) was added to an approximate final amount of 60 g kg–1 and mixed. The water content was maintained at 60% of the maximum water-holding capacity. A box containing the soil was covered with a lid using pine halls and incubated at 25°C. In the analysis, soil samples were taken from 10 distinct points in the box, mixed, and stored at –80°C.
16S rRNA gene analysis of the prokaryotic community structure
DNA was extracted from 0.4 g of each soil sample according to a previously reported method (Takada-Hoshino and Matsumoto, 2004) and dissolved in 80 μL of DNase-free water using a FastDNA SPIN Kit for Soil (MP Biomedicals). PCR was performed on extracted DNAs to amplify the V4 region of the 16S rRNA gene using Ex Taq DNA polymerase (Takara Bio) and the following primers: 515F (5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-GGACTACHVGGGTWTCTAAT-3′) (Caporaso et al., 2011). The PCR products of appropriate sizes were purified using a Wizard SV Gel and PCR Clean-Up System (Promega). PCR fragments (20 ng) were subjected to paired-end sequencing using an Illumina MiSeq platform (Illumina) at Fasmac. Sequences were quality filtered and analyzed with the Quantitative Insights Into Microbial Ecology (QIIME) pipeline (version 1.9.0 [Caporaso et al., 2010]). The remaining sequences (see Table S1 for the detailed numbers of sequences) were clustered into OTUs at the 97% level and classified using the Greengenes 13-8 reference database (Caporaso et al., 2011; McDonald et al., 2012) as implemented in QIIME. Close relatives of representative OTUs were identified by a BLASTn analysis and a phylogenetic analysis using the SILVA database (SILVA SSU Ref NR_128 database) and backbone tree (tree_SSURefNR99_1200slv_128) in the ARB program for the sequence analysis (Ludwig et al., 2004).
Isolation and classification of microorganisms
Upland soil was incubated for 5 and 45 d with powdered chitin (Kumeta et al., 2018). Microbial strains were then isolated from chitin-treated soil by spreading a soil suspension on YG agar plates (0.1% [w/v] glucose, 0.1% [w/v] yeast extract [BD], 0.03% [w/v] dipotassium hydrogen phosphate [K2HPO4], 0.02% [w/v] KH2PO4, 0.01% [w/v] MgSO4, and 1.5% [w/v] agar [pH 6.8]) followed by an incubation at 30°C for 7 d. Microbial strains were purified by plate streaking. Each of the purified strains was cultivated in YG liquid medium, harvested by centrifugation, and subjected to total DNA preparation using a Quick-DNA Bacterial/Fungal kit (Zymoresearch). 16S rRNA genes were amplified by PCR using KOD FX Neo DNA polymerase (TOYOBO) according to the manufacturer’s instructions with the following primers: 27f (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1522r (5′-AAGGAGGTGATCCAGCCGCA-3′). PCR products were electrophoresed, and DNA fragments of approximately 1,500 bp in length were purified using a Wizard SV Gel and PCR Clean-Up System (Promega). The nucleotide sequences of the purified DNA fragments were first partially elucidated using the fragments as templates with the primers 27f (see above for the nucleotide sequence) and 518r (5′-GTATTACCGCGGCTGCTG-3′) in order to identify the strains obtained briefly. Raw data were analyzed using Genetyx software (Nihongenetics), and nucleotide sequences were elucidated. A homology search was performed with Blast (Altschul et al., 1990) at the DNA Data Bank of Japan (DDBJ). The whole sequences of the amplified 16S rRNA gene fragments originating from the Lysobacter strains were elucidated using the purified PCR product as the template. 16S rRNA gene sequences were aligned with default parameters using MEGA v.7.0 software (Kumar et al., 2016). The phylogenetic tree was then constructed with the neighbor-joining method by MEGA v.7.0 software. To evaluate tree topologies, bootstrap values were calculated with 1,000 replicates.
Chitinolytic evaluation of bacterial isolates
Isolated strains were inoculated into minimal medium (MM) (10 mM K2HPO4, 10 mM KH2PO4, 1 mM CaCl2, 0.5 mM MgCl2 supplemented with 0.1% [v/v] trace element solution, and 1.5% [w/v] agar) (Schlochtermeier et al., 1992) containing 0.1% (w/v) colloidal chitin as a carbon source (MMCC medium), as reported previously (Saito et al., 2013) and on MMCC medium supplemented with 0.1% (w/v) yeast extract (YECC medium). Cultures were incubated at 30°C for 10 d. Chitinolytic ability was assessed by the formation of clearing zones around the colonies.
Evaluating auxotrophy in isolated Lysobacter strains
A 1-μL cell suspension of each Lysobacter isolate was spotted so that it formed a line on MM agar medium containing 10 mM glucose as a carbon source. On one side of the spotted line, 7 μL of 2% (w/v) yeast extract, casamino acid, or one of several amino acids (l-alanine, l-arginine, l-asparagine, l-cysteine, d-glutamic acid, l-glutamine, glycine, dl-homocysteine, l-leucine, l-lysine, d-methionine, l-methionine, l-proline, d-serine, l-threonine, or l-valine) was streaked in a linear manner perpendicular to the spotted line (Fig. S1). Cultures were incubated at 30°C for 7 d, and growth was monitored for the indication of auxotrophy. Five type strains of Lysobacter spp. supplied by the Japan Collection of Microorganisms (JCM) (RIKEN, Japan) and the Biological Resource Center, NITE (NBRC, Japan) were used for comparison: L. rhizosperae JCM 30321T, L. panacisoli JCM 19212T, L. niastensis NBRC 106399T, L. koreensis NBRC 101156T, and L. concretionis NBRC 102010T.
PCR primer design for Lysobacter spp.
To detect Lysobacter in incubated soils, we used the previously reported primers: L4 (5′-GAGCCGACGTCGGATTAGCTAGTT-3′) (Hu, 2010), which was designed to detect Lysobacter (Hu, 2010) in soil, and Fw_Lyso_guanxl (non-selective forward primer) (5′-CAACGCGAAGAACCTTACC-3′) and Rev_Lyso_guanxl (selective reverse primer) (5′-TGCAGCACCTGTCTCAC-3′) to analyze the indigenous population of three closely related Lysobacter spp. using real-time PCR with a TaqMan probe (Postma et al., 2011).
To design PCR primers for detecting and quantifying Lysobacter spp., the 16S rRNA gene sequences of the seven isolated Lysobacter strains, as well as 40 strains of Xanthomonadaceae, were aligned to identify DNA regions with nucleotide sequences specific for Lysobacter spp. (Fig. S2). The specificities of the designed primers were confirmed by PCR using Lysobacter spp. total DNA, as described above, with the following strains: Pseudoxanthomonas sp. 45-47-2 and 45-43 (isolated from chitin-treated incubated soil in the present study), P. koreensis NBRC 101160T, P. taiwanensis NBRC 101072T, P. japonensis NBRC 101033T, Xanthomonas campestris NBRC 13551, Stenotrophomonas
nitritireducens JCM 13311T, Dokdonella immobilis JCM 15763T, and D. soli JCM 15421T. PCR was conducted using TaKaRa Ex Taq DNA polymerase Hot Start Version (Takara Bio). Denaturing, annealing, and extension temperatures were set to 94°C for 15 s, 70.5°C for 30 s, and 72°C for 2 min, respectively.
Detection and quantification of Lysobacter spp. in soil
To investigate whether methionine increases the population of Lysobacter, air-dried and sieved Brown Forest soil (see above) was placed in a sterile polypropylene tube, and its water content was adjusted to 60% of its maximum water-holding capacity with sterilized MilliQ water supplemented with or without l-methionine (final 0.1% [w/w]). The same concentration of glucose was added to another soil sample for comparison. After an incubation at 25°C, total DNA was extracted from 0.4 g of each soil sample according to a previously reported method (Takada-Hoshino and Matsumoto, 2004) and dissolved in 80 μL of DNase-free water using a FastDNA SPIN Kit for Soil (MP Biomedicals). Regarding the detection and quantification of Lysobacter spp., PCR was performed using the 16S_Lyso_F1 and 16S_Lyso_R1 primers (see Results and Discussion for details). After end-point PCR, which was performed under the conditions described (see above), the products were electrophoresed on agarose gels and visualized by staining with ethidium bromide. Regarding the quantification of DNA, an Applied Biosystems 7500 Fast Real-Time PCR System (Thermo Fisher Scientific) was used with SYBR Green and TB GreenTM Premix Ex Taq TM II (Takara Bio). Using the total DNA of L. panacisoli JCM 19212T as the template, the corresponding PCR product obtained was used as the standard.
Nucleotide sequence accession numbers
The 16S rRNA gene sequences of the seven Lysobacter isolates, as well as two Pseudoxanthomonas strains isolated in the present study, have been deposited in the DDBJ under accession numbers LC481367-LC481375. Illumina sequencing data were deposited in the DDBJ/ENA/GenBank database under BioProject ID PRJDB8756 and BioSample ID SAMD00185699–SAMD00185712.
Results and Discussion
Bacterial community structure fluctuations in incubated upland soil supplemented with chitin
After incubating chitin-treated soil samples at 25°C for 5 d, approximately 80% of the bacterial community consisted of the genus Streptomyces (Fig. 1), the replication of which markedly increased in chitin-supplemented soil (Mitchell and Alexander, 1962; Sneh and Henis, 1970). As the population of Streptomyces decreased, those of Pseudoxanthomonas and Lysobacter bacteria increased (Fig. 1). Bacteria belonging to the genera Cellulosimicrobium, Streptosporangium, and Nonomuraea also increased following the decrease in the population of Streptomyces. Among the genera with populations that increased in incubated chitin-supplemented upland soil, strains of Streptomyces, Lysobacter, Cellulosimicrobium, and Streptosporangium exhibited chitin-degrading activity and extracellular chitinase production (Deshpande, 1986; Fleuri et al., 2009; Qian et al., 2012; Shirlin and Kalaiarasi, 2016). They also possessed several putative chitinase genes (CAZy: http://www.cazy.org [Henrissat and Davies, 1997]) (Table S2), suggesting the ability to utilize chitin as carbon and nitrogen sources. In contrast, the chitin degradation activities of Nonomuraea and Pseudoxanthomonas strains currently remain unknown. The populations of these genera may utilize chitin degradation products generated by the extracellular chitinases produced by chitinolytic microorganisms. However, genome sequence analyses revealed the presence of putative chitinase genes on the chromosomes of N. gerenzanensis ATCC 39727, N. sp. ATCC 55076, and P. suwonensis J1 (seven, six, and one putative genes, respectively) (CAZy) (Table S2), demonstrating the potential chitinolytic activity of some strains of Nonomuraea and Pseudoxanthomonas.
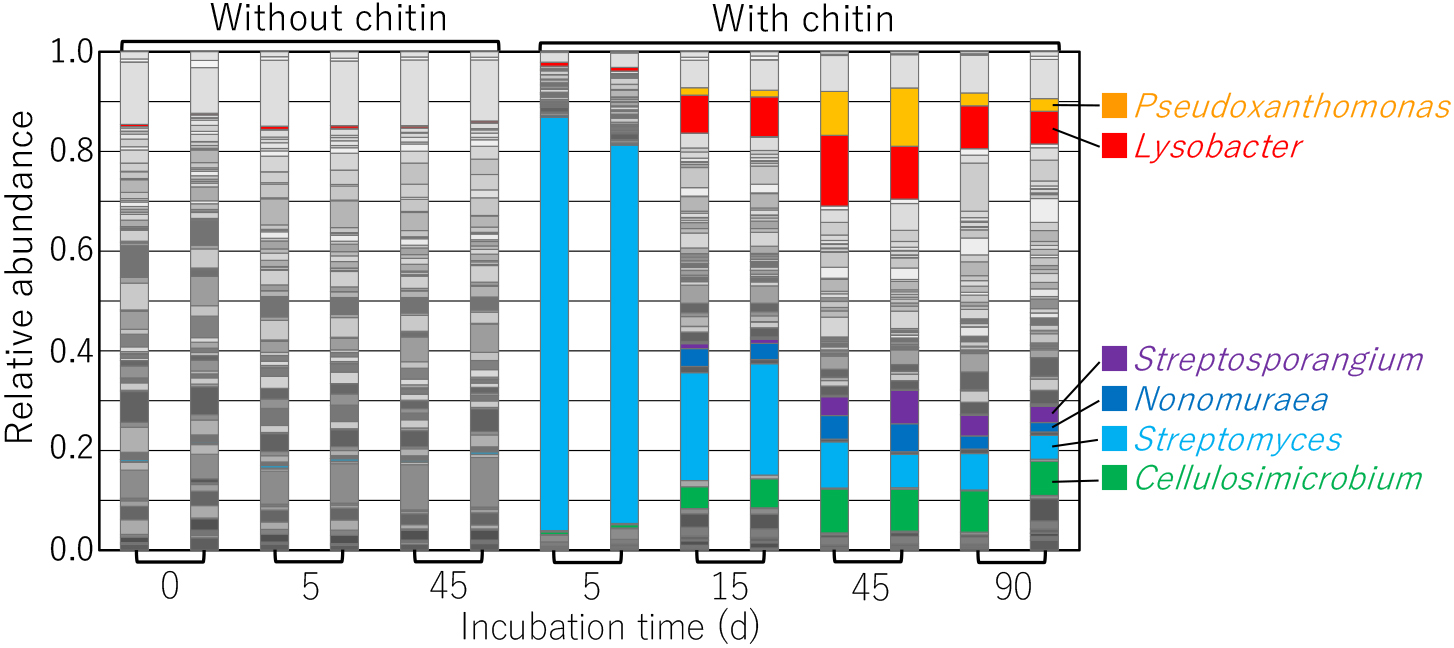
To evaluate the community of culturable chitin-degrading bacteria in chitin-added soil, 83 and 79 (total of 162) strains were isolated, using non-selective agar medium, from chitin-supplemented soils after an incubation at 25°C for 5 and 45 d, respectively. On MMCC agar medium, which contains colloidal chitin as a carbon source, 59 (71%) and 27 (37%) strains isolated from the 5- and 45-day-incubated soil samples, respectively, exhibited chitin degradation. On YECC agar medium, which contains yeast extract in addition to colloidal chitin, 69 (83%) and 35 (45%) strains isolated from the 5- and 45-day-incubated soil samples, respectively, showed chitin degradation. Therefore, the number of chitin-degrading isolates was higher in 5-day-incubated chitin-added soil than in 45-day-incubated soil. This is consistent with previous findings showing that chitinase activity was higher in 5-day-incubated soil than in 45-day-incubated soil (Kumeta et al., 2018). The greater number of chitin-degrading strains observed on YECC than on MMCC suggests that some of the strains are auxotrophs.
Table 1.
Chitinolytic activities of bacterial strains isolated from chitin-treated upland soil.
Incubation time of
chitin-treated upland soil
(d) |
Number of
microbial isolates* |
Number of strains exhibiting
chitinolytic activity
(% against the number of isolates) |
| on MMCC** |
On YECC** |
| 5 |
83 (100) |
59 (71) |
69 (83) |
| 45 |
79 (100) |
27 (37) |
35 (45) |
* Microbial strains were isolated using YG agar medium containing yeast extract and glucose (see Materials and Methods for a detailed composition).
** MMCC, minimal agar medium containing 0.1% (w/v) colloidal chitin as a carbon source; YECC, MMCC supplemented with 0.1% (w/v) yeast extract.
Partial 16S rRNA gene sequences were elucidated for 161 out of the 169 strains isolated from chitin-added incubated upland soil using a non-selective medium. Seven of the identified strains belonged to the genus Lysobacter. Six out of the seven Lysobacter strains exhibited chitin-degrading activity on YECC agar medium, suggesting the involvement of chitinolytic Lysobacter strains in chitin degradation in incubated upland soil. One strain (5-21a) was isolated from 5-day-incubated soil, while the other 6 strains were obtained from 45-day-incubated soil. In contrast, 46 and 9 chitinolytic strains isolated from 5- and 45-day-incubated soil were identified as Streptomyces (unpublished data). These results were consistent with those of the bacterial community structure analysis showing that the numbers of Lysobacter and Streptomyces bacteria were higher and lower, respectively, in 45-day-incubated soil than in 5-day-incubated soil (Fig. 1). Amplified 16S rRNA gene sequences (approximately 1.4 kb) from the seven Lysobacter isolates revealed that 5 strains (5-21a, 45-18, 45-27, 45-29, and 45-65) were closely related to L. soli (99.3–100%) (Srinivasan et al., 2010), strain 45-28 was closely related to L. niastensis NBRC106399T (99.0%) (Weon et al., 2007), and strain 45-72 was closely related to L. dokdonensis DS-58 (99.9%) (Oh et al., 2011). Strain 45-72, which did not exhibit chitinolytic activity, was phylogenetically apart from the other six Lysobacter isolates (Fig. 2). The six chitinolytic Lysobacter strains did not significantly grow on MMCC agar medium, but still grew well, indicating chitinolytic activity. Since we suspected auxotrophy, we investigated the effects of nutritional characteristics on the growth of the strains.
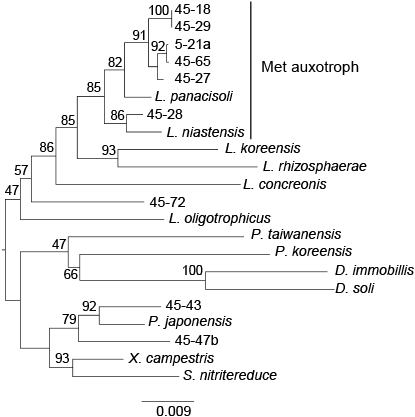
Lysobacter sp. strain 5-21a isolated from incubated chitin-supplemented soil did not significantly grow on minimal media supplemented with colloidal chitin or glucose as a carbon source. However, it grew in the presence of yeast extract or casamino acid (Fig. S1 and Table 2). These results suggest that Lysobacter sp. 5-21a is an amino acid auxotroph. Among the 16 amino acids tested, only l- or d-methionine (Met) rescued the growth of 5-21a on minimal agar medium supplemented with glucose (Table 2), demonstrating Met auxotrophy. Among the six other isolates of Lysobacter, five strains (45-18, 45-27, 45-28, 45-29, and 45-65) also demonstrated Met auxotrophy, while one (45-72) did not (Table 2). 45-72, which is phylogenetically apart from the other six Lysobacter strains isolated in the present study (Fig. 2), may be an auxotroph against another amino acid. Among the five type strains of Lysobacter tested, L. panacisoli and L. niastensis, which are closely related to the five Met auxotroph Lysobacter isolates (Fig. 2), also indicated Met auxotrophy, while the three other species (L. rhizospherae, L. koreensis, and L. concretionis) did not (Table 2). The growth of L. rhizosphaerae, similar to Lysobacter sp. 45-72, was enhanced by casamino acid, but not by Met, and casamino acid did not enhance the growth of L. koreensis or L. concretionis. These results indicate that Met auxotrophy observed in the Lysobacter isolates was shared with some type strains of Lysobacter spp.
Table 2.
Effects of amino acids on the growth of
Lysobacter strains on minimal agar medium supplemented with glucose.
|
Isolates of Lysobacter spp. |
|
Type strains of Lysobacter spp. |
| 5-21a |
45-18,
45-27,
45-28,
45-29,
4-65 |
45-72 |
L. panacisoli
JCM 19212T |
L. niastensis
NBRC 106399T |
L. rhizospherae
JCM 30321T |
L. koreensis
NBRC 101156T |
L. concretionis
NBRC 102010T |
| Casamino acid |
+ |
+ |
+ |
|
+ |
+ |
+ |
– |
– |
| l-Met |
+ |
+ |
– |
|
+ |
+ |
– |
– |
– |
| d-Met |
+ |
+ |
– |
|
+ |
+ |
– |
– |
– |
| Other amino acids* |
– |
nt |
nt |
|
nt |
nt |
nt |
nt |
nt |
* l-alanine, l-arginine, l-asparagine, l-cysteine, d-glutamic acid, l-glutamine, glycine, d,l-homocysteine, l-leucine, l-lysine, l-proline, d-serine, l-threonine, or l-valine.
+, growth was significantly enhanced; –, growth was not significantly enhanced; nt, not tested.
The growth of Lysobacter sp. 5-21a was not rescued by cysteine or homocysteine (Table 2). Met auxotroph Lysobacter strains were not able to utilize l-Met or d-Met as a sole carbon source (data not shown). 5-21a does not appear to have the capacity to synthesize or metabolize Met via these amino acids and may use Met only for anabolism, such as protein synthesis. It is important to note that Met auxotroph Lysobacter strains, including two type strains, utilize d-Met as well as l-Met (Table 2); however, the purity of commercially obtained d-Met needs to be examined. Pseudomonas putida IFO 12996 produces broad specificity amino acid racemase (BAR) (E.C. 5.1.1.10) (Soda and Osumi, 1969). Vibrio cholerae also produces a periplasmic broad spectrum racemase (BsrV) that racemizes several types of amino acids (Lam et al., 2009; Espaillat et al., 2014). BAR and BsrV racemize d-Met and other amino acids (Kino et al., 2007; Espaillat et al., 2014). Lysobacter strains may produce these types of enzymes to utilize d-Met as a Met source.
PCR primer design for Lysobacter spp.
In order to select PCR primers for investigating the effects of Met treatments on the population of Lysobacter in soil, we initially demonstrated the specificities of the primers reported previously, and found that the primer set L4 (Hu, 2010) and another universal bacterial primer 518R, as well as Fw_Lyso_guanxl and Rev_Lyso_guanxl (Postma et al., 2011), amplified the corresponding DNA fragment from the total DNAs of Lysobacter spp. and strains from the genus Pseudoxanthomonas (Table 3. See Fig. 2 for their phylogenetic positions). Therefore, we designed a new primer set (16S_Lyso_F1 [5′-CGGGTTGTAAAGCWCTTTTGTCC-3′] and 16S_Lyso_R1 [5′-GAAGTTAGCCGGTGCTTATTCTTCC-3′]) based on the alignment of 16S rRNA genes (Fig. S2) that was more specific against Lysobacter. The new primer set successfully amplified DNA fragments of the expected sizes in all Lysobacter strains tested, except for L. concretionis (Fig. 3), the nucleotide sequence of which does not match the 3′-end of 16S_Lys_F1 (Fig. S2). In contrast, the primer set did not amplify the 16S rRNA gene from closely related genera including Pseudoxanthomonas, Xanthomonas, and Stenotrophomonas when using total DNA. However, the primer set also amplified the corresponding DNA fragment from the total DNA of D. soli, the nucleotide sequence of which matches those of the primers. However, it did not amplify DNA fragments from D. immobilis (Fig. 3 and S2). Thus, we concluded that the designed primer set was more specific for detecting Lysobacter spp. than those reported to date, and also amplified the corresponding gene of at least D. soli.
Table 3.
Amplification of target DNA fragments
| Species |
Strain |
Primers (References) |
L4 (Hu, 2010)
518r |
Fw_Lyso_guanxl
Rev_Lyso_guanxl
(Postma et al., 2011) |
16S_Lyso_F1
16S_Lyso_R1
(This study) |
| L. koreensis |
NBRC101156T |
+ |
– |
+ |
| L. rhizosphaerae |
JCM30321T |
+ |
– |
+ |
| L. concretionis |
NBRC102010T |
+ |
+ |
– |
| L. niastensis |
NBRC106399T |
+ |
+ |
+ |
| L. panacisoli |
JCM19212T |
+ |
+ |
+ |
| L. oligotrophicus |
JCM18257T |
+ |
+ |
+ |
| Lysobacter sp. |
5-21a* |
+ |
+ |
+ |
|
45-65* |
+ |
+ |
+ |
|
45-27* |
+ |
+ |
+ |
|
45-18* |
+ |
+ |
+ |
|
45-29* |
+ |
+ |
+ |
|
45-28* |
+ |
+ |
+ |
|
45-72* |
+ |
+ |
+ |
| Pseudoxanthomonas sp. |
45-43* |
+ |
– |
– |
| Pseudoxanthomonas sp. |
45-47b* |
+ |
+ |
– |
| Pseudoxanthomonas japonensis |
NBRC101033T |
nt |
nt |
– |
| Pseudoxanthomonas koreensis |
NBRC101160T |
nt |
nt |
– |
| Pseudoxanthomonas taiwanensis |
NBRC101072T |
nt |
nt |
– |
| Stenotrophomonas nitritireducens |
JCM13311T |
nt |
nt |
– |
| Xanthomonas campestris |
NBRC13551 |
nt |
nt |
– |
| Dokdonella immobilis |
JCM15763T |
nt |
nt |
– |
| Dokdonella soli |
JCM15421T |
nt |
nt |
+ |
* Strains isolated in this study.
+, amplified; –, not amplified; nt, not tested.

The ability of the PCR primer set to evaluate the population of Lysobacter was confirmed by both semi-quantitative (normal) PCR and quantitative PCR (qPCR) using DNA samples prepared for the amplicon sequencing analysis (Fig. 1) as templates. Normal PCR results were consistent with the amplicon sequencing analysis of Lysobacter populations (i.e. the amount of the PCR product was lower in soil without added chitin, but increased in chitin-supplemented soil) (Fig. 4). Therefore, a positive correlation appears to exist between the amount of the PCR product and the amount of Lysobacter in chitin-supplemented soil. The results of the qPCR analysis using SYBR green were also consistent with the amplicon sequencing analysis, indicating the utility of the primer set at quantifying Lysobacter populations (Fig. 4). Thus, we concluded that the primers 16S_Lyso_F1 and 16S_Lyso_R1 are appropriate for evaluating the quantity of Lysobacter species, even though they also amplify the corresponding gene in some species of the genus Dokdonella.
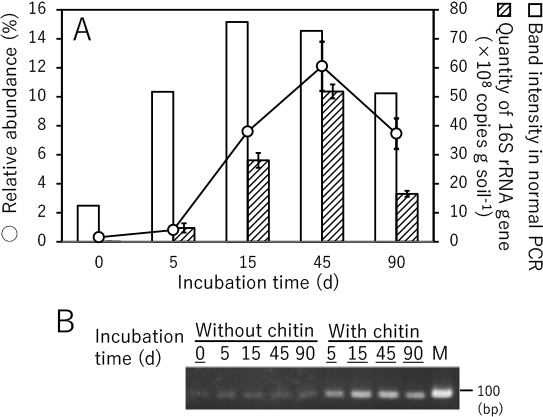
The amount of the PCR product amplified with the 16S_Lyso_F1 and 16S_Lyso_R1 primers increased when DNA templates prepared from incubated upland soil supplemented with 0.1% d,l-Met were examined; however, it did not increase in non-supplemented soil or soil supplemented with 0.2% glucose (Fig. 5). These results strongly suggest that the population of Lysobacter increased due to the addition of d,l-Met. An increase in the Lysobacter spp. population by Met was already observed on a partial-nitritation biofilter for wastewater treatment (Gonzalez-Martinez et al., 2016). The findings obtained suggested that Lysobacter spp. does not grow due to methionine consumption itself, but due to predation of the bacterial biomass in the system (Gonzalez-Martinez et al., 2016) because Lysobacter spp. has been identified as heterotrophic with lytic activity against other microorganisms, including fungi and nematodes (Christensen and Cook, 1978). The present results suggesting the Met auxotrophy of Lysobacter strains imply that the increased Lysobacter spp. population on the partial nitritation biofilter may be a Met auxotroph.

Lysobacter spp. were isolated from clay soils and not from sandy soils found in the arable fields of organic farms in the Netherlands (Postma et al., 2008). A real-time PCR analysis demonstrated that populations of L. antibioticus, L. capsica, and L. gummosus were approximately 6.22–6.95 log gene copies g of soil–1 larger in clay soils from arable fields of organic farms than in sandy soils (Postma et al., 2011). We herein used light clay soil from an upland field in Japan (see Materials and Methods for details). Met may be useful for increasing and/or activating indigenous Lysobacter spp. in soils; however, the presence of Lysobacter spp. and its growth modulation by chitin or Met need to be investigated in soils of different textures.
In the present study, we demonstrated an immediate increase and subsequent decrease in Streptomyces (Fig. 1), one of the main decomposers of chitin in soil. As the population of Streptomyces decreased over time, Lysobacter, Pseudoxanthomonas, Cellulosimicrobium, Streptosporangium, and Nonomuraea populations increased (Fig. 1). We isolated six Lysobacter strains exhibiting chitin-degradation activity from chitin-supplemented soil, suggesting that Lysobacter was involved in chitin degradation in chitin-treated upland soil. Lysobacter isolates were Met auxotrophs. The population of Lysobacter increased in Met-treated incubated soils (Fig. 5). We assume that Met is a factor contributing to increases in the population of Lysobacter in chitin-added upland soil. In a study using synthetic syntrophic microbial communities, biosynthetically costly amino acids, including Met, lysine, isoleucine, arginine, and aromatics, were found to more likely promote stronger cooperative interactions than amino acids that are cheaper to produce (Mee et al., 2014). Met may be a key compound that triggers the transition from Streptomyces to Lysobacter, and possibly other bacterial genera in chitin-treated upland soil, as the dominant bacterial population. This hypothesis is currently being investigated.
Acknowledgements
The authors would like to thank Daichi Homma, Akihiro Ohta, and Naoya Usami at SIST for their assistance. This study was supported, in part, by KAKENHI grants 25450086 (2013–2015) and 16K07648 (2016–2019).
References
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. (1990) Basic local alignment search tool. J Mol Biol
215: 403–410.
- Buxton, E.W., Khalifa, O., and Ward, V. (1965) Effect of soil amendment with chitin on pea wilt caused by Fusarium oxysporum f. pisi. Ann Appl Biol
55: 83–88.
- Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K., et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods
7: 335–336.
- Caporaso, J.G., Lauber, C.L., Walters, W.A., Berg-lyons, D., Lozupone, C.A., Turnbaugh, P.J., et al. (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A
108: 4516–4522.
- Christensen, P., and Cook, F.D. (1978)
Lysobacter, a new genus of nonfruiting, gliding bacteria with a high base ratio. Int J Syst Evol Microbiol
28: 367–393.
- Cretoiu, M.S., Korthals, G.W., Visser, J.H.M., and van Elsas, J.D. (2013) Chitin amendment increases soil suppressiveness toward plant pathogens and modulates the actinobacterial and oxalobacteraceal communities in an experimental agricultural field. Appl Environ Microbiol
79: 5291–5301.
- Deshpande, M.V. (1986) Enzymatic degradation of chitin and its biological applications. J Sci Ind Res (India) 45: 273–281.
- Espaillat, A., Carrasco-Lόpez, C., Bernardo-García, N., Pietrosemoli, N., Otero, L.H., Alvarez, L., et al. (2014) Structural basis for the broad specificity of a new family of amino-acid racemases. Acta Crystallogr D Biol Crystallogr
70: 79–90.
- Fleuri, L.F., Kawaguti, H.Y., and Sato, H.H. (2009) Production, purification, and application of extracellular chitinase from Cellulosimicrobium cellulans 191. Braz J Microbiol
40: 623–630.
- Gonzalez-Martinez, A., Rodoriguez-Sanchez, A., Garcia-Ruiz, M.J., Osorio, F., and Gonzalez-Lopez, J. (2016) Impact of methionine on a partial-nitritation biofilter. Environ Sci Pollut Res
23: 6651–6660.
- Gooday, G.W. (1991) Chitinases. In Enzymes in Biomass Conversion. Latham, G.F., and Himmel, M.E. (eds). Washington DC: American Chemical Society, pp. 478–485.
- Henis, Y., Sneh, B., and Katan, J. (1967) Effect of organic amendments on Rhizoctonia and accompanying microflora in soil. Can J Microbiol
13: 643–650.
- Henrissat, B., and Davies, G.J. (1997) Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol
7: 637–644.
- Hu, Y. (2010) Detection methods for the genus Lysobacter and the species Lysobacter enzymogenes. PhD Thesis, University of Nebraska.
- Jacquiod, S., Franqueville, L., Cécillon, S., Vogel, T.M., and Simonet, P. (2013) Soil bacterial community shifts after chitin enrichment: An integrative metagenomic approach. PLoS One
8: e79699.
- Khalifa, O. (1965) Biological control of Fusarium wilt of peas by organic soil amendments. Ann Appl Biol
56: 129–137.
- Kino, K., Sato, M., Yoneyama, M., and Kimura, K. (2007) Synthesis of DL-tryptophan by modified broad specificity amino acid racemase from Pseudomonas putida IFO 12996. Appl Microbiol Biotechnol
73: 1299–1305.
- Kumar, S., Stecher, G., and Tamura, K. (2016) MEGA7: Molecular evolutionary genetics analysis version7.0 for bigger datasets. Mol Biol Evol
33: 1870–1874.
- Kumeta, Y., Inami, K., Ishimaru, K., Yamazaki, Y., Sameshima-Saito, R., and Saito, A. (2018) Thermogravimetric evaluation of chitin degradation in soil: implication for the enhancement of ammonification of native organic nitrogen by chitin addition. Soil Sci Plant Nutr
64: 512–519.
- Lam, H., Oh, D.C., Cava, F., Takacs, C.N., Clardy, J., de Pedro, M.A., and Waldor, M.K. (2009)
d-amino acids govern stationary phase cell wall remodeling in bacteria. Science
325: 1552–1555.
- Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, et al. (2004) ARB: a software environment for sequence data. Nucleic Acids Res
32: 1363–1371.
- McDonald, D., Price, M.N., Goodrich, J., Nawrocki, E.P., DeSantis, T.Z., Probst, A., et al. (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J
6: 610–618.
- Mee, M.T., Collins, J.J., Church, G.M., and Wang, H.H. (2014) Syntrophic exchange in synthetic microbial communities. Proc Natl Acad Sci U S A
111: E2149–E2156.
- Mendes, R., Kruijt, M., de Bruijn, I., Dekkers, E., van der Voort, M., Schneider, J.H., et al. (2011) Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science
332: 1097–1100.
- Mian, I.H., Godoy, G., Shelby, R.A., Rodriguez-Kabana, R., and Morgan-Jones, G. (1982) Chitin amendments for control of Meloidogyne arenaria in infested soil. Nematropica
12: 71–84.
- Mitchell, R., and Alexander, M. (1962) Microbiological processes associated with the use of chitin for biological control. Soil Sci Soc Am Proc
26: 556–558.
- Murakami, H., Kuroyanagi, Y., Tsushima, S., and Shishido, Y. (2009) Effects of application of organic materials on the development of clubroot disease caused by Plasmodiophora brassicae. Soil Microorganisms
63: 3–8 (in Japanese).
- Muzzarelli, R.A. (1977) Chitin. New York, NY: Pergamon Press.
- Nicol, S. (1991) Life after death for empty shells. New Sci
129: 46–48.
- Oh, K.H., Kang, S.J., Jung, Y.T., Oh, T.K., and Yoon, J.H. (2011)
Lysobacter dokdonensis sp. nov., isolated from soil. Int J Syst Evol Microbiol
61: 1089–1093.
- Postma, J., Schilder, M.T., Bloem, J., and van Leeuwen-Haagsma, W.K. (2008) Soil suppressiveness and functional diversity of the soil microflora in organic farming systems. Soil Biol Biochem
40: 2394–2406.
- Postma, J., Schilder, M.T., and van Hoof, R.A. (2011) Indigenous populations of three closely related Lysobacter spp. in agricultural soils using real-time PCR. Microb Ecol
62: 948–958.
- Qian, G., Wang, Y., Qian, D., Fan, J., Hu, B., and Liu, F. (2012) Selection of available suicide vectors for gene mutagenesis using chiA (a chitinase encoding gene) as a new reporter and primary functional analysis of chiA in Lysobacter enzymogenes strain OH11. World J Microbiol Biotechnol
28: 549–557.
- Saito, A., Ebise, H., Orihara, Y., Murakami, S., Sano, Y., Kimura, A., et al. (2013) Enzymatic and genetic characterization of the DasD protein possessing N-acetyl-β-d-glucosaminidase activity in Streptomyces coelicolor A3(2). FEMS Microbiol Lett
340: 33–40.
- Sarathchandra, S.U., Watson, R.N., Cox, N.R., di Menna, M.E., Brown, J.A., Burch, G., and Neville, F.J. (1996) Effects of chitin amendment of soil on microorganisms, nematodes, and growth of white clover (Trifolium repens L.) and perennial ryegrass (Lolium perenne L.). Biol Fertil Soils
22: 221–226.
- Schlochtermeier, A., Walter, S., Schröder, J., Moorman, M., and Schrempf, H. (1992) The gene encoding the cellulase (Avicelase) Cel1 from Streptomyces reticuli and analysis of protein domains. Mol Microbiol
6: 3611–3621.
- Shirlin, J.M., and Kalaiarasi, J.M.V. (2016) Isolation of novel marine chitinolytic actinomycete Streptosporangium sp. and production optimization of extracellular chitinase. Int J Sci Res Sci Technol
2: 311–316.
- Sneh, B., and Henis, Y. (1970) Production of antifungal substances active against Rhizoctonia solani in chitin-amended soil. Phytopathology
62: 595–600.
- Soda, M., and Osumi, T. (1969) Crystalline amino acid racemase with low substrate specificity. Biochem Biophys Res Commun
35: 363–368.
- Srinivasan, S., Kim, M.K., Sathiyaraj, G., Kim, H.B., Kim, Y.J., and Yang, D.C. (2010)
Lysobacter soli sp. nov., isolated from soil of a ginseng field. Int J Syst Evol Microbiol
60: 1543–1547.
- Takada-Hoshino, Y., and Matsumoto, N. (2004) An improved DNA extraction method using skim milk from soils that strongly adsorb DNA. Microbes Environ
19: 13–19.
- Van Eck, W.H. (1978) Autolysis of chlamydospores of Fusarium solani f. sp. cucurbitae in chitin and laminarin amended soils. Soil Biol Biochem
10: 89–92.
- Veldkamp, H. (1955) A study of the aerobic decomposition of chitin by microorganisms. Madedel Landbouwhogeschool Wageningen
55: 127–174.
- Weller, D.M., Raaijmakers, J.M., McSpadden, B.B., and Thomashow, L.S. (2002) Microbial populations responsible for specific soil suppressiveness to plant pathogen. Annu Rev Phytopathol
40: 309–348.
- Weon, H.W., Kim, B.Y., Kim, M.K., Yoo, S.H., Kwon, S.W., Go, S.J., and Stackebrandt, E. (2007)
Lysobacter niabensis sp. nov. and Lysobacter niastensis sp. nov., isolated from green house soils in Korea. Int J Syst Evol Microbiol
57: 548–551.
- Wieczoreck, A.S., Hetz, S.A., and Kolb, S. (2014) Microbial responses to chitin and chitosan in oxic and anoxic agricultural soil slurries. Biogeoscience
11: 3339–3352.