Abstract
The genome of Streptomyces scabies, the predominant causal agent of potato common scab, encodes a potential cutinase, the protein Sub1, which was previously shown to be specifically induced in the presence of suberin. The sub1 gene was expressed in Escherichia coli and the recombinant protein Sub1 was purified and characterized. The enzyme was shown to be versatile because it hydrolyzes a number of natural and synthetic substrates. Sub1 hydrolyzed p-nitrophenyl esters, with the hydrolysis of those harboring short carbon chains being the most effective. The Vmax and Km values of Sub1 for p-nitrophenyl butyrate were 2.36 mol g–1 min–1 and 5.7 10–4 M, respectively. Sub1 hydrolyzed the recalcitrant polymers cutin and suberin because the release of fatty acids from these substrates was observed following the incubation of the enzyme with these polymers. Furthermore, the hydrolyzing activity of the esterase Sub1 on the synthetic polymer polyethylene terephthalate (PET) was demonstrated by the release of terephthalic acid (TA). Sub1 activity on PET was markedly enhanced by the addition of Triton and was shown to be stable at 37°C for at least 20 d.
Streptomycetes are Gram-positive bacteria that are known for their ability to produce a wide range of secondary metabolites and for the complexity of their morphological development. Although most streptomycetes species are saprophytic soil inhabitants, some are plant pathogens. Among them, Streptomyces scabies is the predominant causal agent of potato common scab and causes important economic losses in Canada (Hill and Lazarovits, 2005), as well as in most potato growing areas. Common scab is characterized by corky lesions on the surface of potato tubers. Similar to other soil-inhabiting streptomycetes, S. scabies produces a large variety of extracellular enzymes, including various glycosyl hydrolases and esterases (Komeil et al., 2013; Beaulieu et al., 2016). These enzymes may participate in pathogenesis because the penetration of S. scabies into host plants is considered to be facilitated by the secretion of extracellular cell wall-degrading enzymes (Beauséjour et al., 1999).
The potato tuber is covered by a periderm that is composed of three types of tissues: phellem, phellogen, and phelloderm (Tyner et al., 1997). The wall of phellem cells impregnated with suberin, a plant polymer recalcitrant to bio-degradation, is composed of a polyaromatic domain covalently linked to a polyaliphatic moiety (Bernards, 2002). The polyaromatic domain, a lignin-like structure, consists of a hydroxycinnamic acid-derived polymeric matrix (Bernards and Lewis, 1998). The polyaliphatic domain shares structural and chemical similarities with cutin, another polyester component of plant cuticles. Cutin and suberin both act as physical barriers against plant pathogens (Khatri et al., 2011). Cutin and suberin are polymers of fatty acid derivatives linked by ester bonds. Cutin is mostly composed of C16 and C18 ω-hydroxyacids, polyhydroxyacids, epoxyacids, and α,ω-dicarboxylic acids. Suberin may be distinguished from cutin by higher contents of hydroxycinnamic acids, fatty alcohols, and saturated aliphatics with long chains (Beisson et al., 2012).
Cutinases hydrolyze the plant leaf cuticle by cleaving the ester bounds of cutin (Dutta et al., 2009). Therefore, cutinases belong to the esterase group, and more specifically to a class of serine esterases that contain the catalytic triad (serine, histidine and aspartate) with the active serine in the consensus sequence Gly-His/Tyr-Ser-X-Gly (Martinez et al., 1994). Some fungal cutinases, such as the cutinase CcCUT1 of the fungus Coprinopsis cinerea, also exhibit the ability to degrade suberin (Kontkanen et al., 2009). As lipolytic enzymes, cutinases have interesting properties for applications in various industrial processes (Carvalho et al., 1999). For example, some cutinases exhibit the ability to degrade synthetic polyesters, such as polycaprolactone (Murphy et al., 1996) and polyethylene terephthalate (PET) (Vertommen et al., 2005; Eberl et al., 2009; Ribitsch et al., 2015).
Previous studies suggested that the bacterium S. scabies possesses the ability to degrade suberin. This pathogenic bacterium exhibits strong esterase activity in the presence of suberized tissues (Beauséjour et al., 1999). Furthermore, a secretome analysis of S. scabies cultures grown in the presence of suberin revealed the presence of esterases, which are predicted to play a role in lipid metabolism (Komeil et al., 2014; Beaulieu et al., 2016). Among them, the Sub1 protein exhibits 33% identity with the cutinase CcCUT1 of the fungus C. cinerea (Komeil et al., 2013). Interestingly, sub1 gene expression was previously reported to be specifically induced in the presence of suberin (Komeil et al., 2013). The cutinase from Aspergillus oryzae (PDB ID: 3GBS) is hallmarked by a central β-sheet of five parallel strands surrounded by ten α-helices (Liu et al., 2009), as found for the predicted three-dimensional structure of the protein Sub1 (Supplemental Fig. S1). The model of the Sub1 protein also predicts the formation of two disulfide bonds (Cys31–Cys103; Cys178–Cys185) and a catalytic triad including residues Ser 114, Asp 182, and His 195 (Komeil et al., 2013).
The main objectives of the present study were to produce the Sub1 protein, purify and characterize its enzymatic properties, and demonstrate that it functions as a polyesterase with the ability to degrade biopolymers, such as cutin and suberin, as well as the synthetic polyester PET.
Materials and Methods
Bacterial strains and culture conditions
An inoculum of S. scabies EF-35 (HER1481) was prepared in tryptic soy broth (108 spores in 25 mL), as described previously (Komeil et al., 2013). Cultures of S. scabies EF-35 were incubated with shaking (250 rpm) at 30°C. Escherichia coli strains DH5α (Invitrogen) and SHuffle T7 (New England Biolabs) were grown in LB medium supplemented where necessary with kanamycin (30 μg mL–1) and were then incubated with shaking (250 rpm) at 37°C.
Suberin and cutin preparation
A suberin-enriched potato periderm was obtained as previously described (Kolattukudy and Agrawal, 1974). The extracted material was dried under a hood, ground using a coffee mill, and stored at room temperature. To further remove residual polysaccharides in the potato periderm, this material was exposed to microbial degradation in the presence of S. scabies EF-35, as described by Beaulieu et al. (2016). The S. scabies inoculum (1 mL) was added to 50 mL of minimal medium consisting of a mineral solution (0.5 g L–1 [NH4]2SO4, 0.5 g L–1 K2HPO4, 0.2 g L–1 MgSO4-7H2O, and 10 mg L–1 FeSO4-7H2O) and 1 g L–1 of the suberin-enriched potato periderm. After a 30-d incubation, 10 mL of fresh mineral solution and 200 μL of the S. scabies inoculum were both added to the culture and the incubation was extended for an additional 30 d. The culture was centrifuged at 3,450×g for 20 min, and the pellet was resuspended in 100 mL of sterile water and then autoclaved for 15 min. The suspension was washed with sterile water to remove bacterial cell debris. The resulting material (purified potato suberin) was dried at 50°C for 24 h. Cutin was isolated from apples following the protocol of Walton and Kolattukudy (1972).
DNA extraction
Genomic DNA was isolated from 48-h bacterial cultures of S. scabies EF-35 using the GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich) according to the manufacturer’s instructions. Plasmid DNA was isolated from 12-h E. coli cultures using the GenEluteTM Plasmid Miniprep Kit (Sigma-Aldrich) following the manufacturer’s instructions.
Cloning of sub1 in E. coli
The sub1 coding sequence, deprived of its signal peptide (GenBank accession number MK689853), was amplified by PCR from the genomic DNA of S. scabies EF-35 using the primers F-pET (5′-ATATCCATGGCCGCCTGCACGGACATCG-3′) and R-pET (5′-ATATCTCGAGTTAGATCTTGGTCGCGGCGAAGG-3′). The PCR mix contained 20 ng of DNA, 2.5 μL of Taq polymerase buffer, 0.5 μL of dNTPs (10 mM), 0.5 μL (each) of forward and reverse primers (10 μM), and 0.125 μL of DNA Taq polymerase (New England Biolabs), in a total volume of 25 μL. PCR conditions consisted of 2 min at 95°C followed by 30 cycles at 95°C for 30 s, at 64°C for 1 min, and at 68°C for 1 min, with a final extension at 68°C for 5 min. PCR was performed using the thermocycler T100 (Bio-Rad). Amplification products were migrated on a 1% agarose gel (Sambrook and Russell, 2001), purified from gels using the MinElute Gel Extraction Kit (Qiagen), and cloned into the pET-30a(+) vector (Novagen). The amplification product and cloning vector pET-30a(+) were both digested using the restriction enzymes NcoI and XhoI. Enzyme T4 DNA ligase (New England Biolabs) was used to ligate plasmid ends to amplicons following the manufacturer’s instructions. Ligation products were heat shock-transformed into competent cells of E. coli DH5α as per the manufacturer’s instructions (New England Biolabs). Bacteria were then incubated overnight on LB agar medium supplemented with kanamycin (30 μg mL–1). The plasmid insert was sequenced at a sequencing and genome genotyping platform (CHUL, University Laval, Quebec City, Canada). The plasmid pET-30a(+), with or without the sub1 insert, was transformed into the expression host E. coli SHuffle T7, as previously described.
Protein extraction
Cultures of E. coli SHuffle T7 carrying pET without or with the sub1 gene insert (E. coli SHuffle T7-pET and E. coli SHuffle T7-pET-sub1) were incubated on LB agar medium supplemented with kanamycin. When OD600 reached 0.6–0.8, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to the culture (0 to 1.0 mM, final concentration) and bacteria were incubated at 25°C for an additional 24 h. Cells were harvested by centrifugation (3,450×g) for 10 min, pellets were washed twice with saline (NaCl 0.9%) and then resuspended in a buffer solution (50 mM NaH2PO4 and 300 mM NaCl, pH 8.0) supplemented with EDTA (2.5 mM). The suspensions were sonicated on ice four times for 10 s and centrifuged (3,450×g) at 4°C for 30 min to remove cell debris. The supernatant was collected and successively passed through filters with pore sizes of 0.45 and 0.2 μm. The resulting protein solution was stored at 4°C.
Purification of the protein Sub1
An affinity column Ni-NT cOmplete His-Tag purification column (Roche) was used to purify the protein Sub1 from the cytoplasmic fraction of E. coli SHuffle T7-pET-sub1, following the manufacturer’s instructions. Elution buffer A (50 mM NaH2PO4, 300 mM NaCl, pH 8.0), supplemented with different concentrations of imidazole (5 to 250 mM), was used for column washing and His6-tagged protein elution. Protein fractions were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) migration along with the marker PageRuler™ Prestained Protein Ladder (Thermo Scientific), as described by Komeil et al. (2014). Proteins were stained with Coomassie brilliant blue R-250 (Bio-Rad; Lauzier et al., 2008) and fractions containing purified Sub1 were pooled. This mixture was dialyzed in phosphate-buffered solution (PBS) to remove imidazole. Protein concentrations were measured according to Bradford (Bradford, 1976).
Esterase activity of Sub1 on p-nitrophenyl esters
Esterase activity was assessed by spectrophotometrically measuring the absorbance of p-nitrophenol using the substrates p-nitrophenyl butyrate (C4), p-nitrophenyl octanoate (C8), p-nitrophenyl decanoate (C10), and p-nitrophenyl dodecanoate (C12) (Sigma-Aldrich). The molar extinction coefficient of p-nitrophenol in Tris-HCl (20 mM, pH 7.5) at room temperature is 12,000 M–1 cm–1 at 420 nm. This enzymatic assay was performed as described previously (Komeil et al., 2013) with slight modifications. In a 1.5-mL plastic cuvette, 20 μL of 100×-diluted Sub1 (60 ng) was added to 970 μL of Tris-HCl (20 mM, pH 7.5), with or without Triton X-100 (0.5%), and 10 μL of a 20 mM p-nitrophenyl ester substrate (0.2 mM final concentration). The absorbance at 420 nm of this reaction mix was measured at room temperature every 10 s for 1 min. The increase in absorbance of each sample was read against a blank without purified protein. One unit (U) was the amount of enzyme liberating 1 μmol of p-nitrophenol min–1 under the assay conditions. The Vmax and Km values of the enzyme Sub1 were assessed using the software GraphPad Prism7, according to the Michaelis-Menten equation, with different concentrations of the C4 substrate.
Esterase activity of Sub1 on natural and synthetic polymers
Suberin and cutin were exposed to the enzyme Sub1 as follows. Suberin or cutin (10 mg) was added to 350 μL of Tris-HCl (20 mM, pH 7.5) supplemented with 50 μL of the purified enzyme Sub1 (15 μg). Control assays (blanks) were made of suberin and cutin without the addition of the enzyme Sub1. The mixture was incubated at room temperature for 20 d. Colorimetric assays of free fatty acids released from the biopolymer were performed every 5 d using a Free Fatty Acid Quantification Colorimetric/Fluorometric Kit (BioVision) according to the manufacturer’s instructions. A standard curve was prepared with palmitic acid to convert absorbance at 570 nm into fatty acid concentrations.
The hydrolyzing activity of Sub1 was also estimated on polyethylene terephthalate (PET) by measuring the amount of terephthalic acid (TA) released from PET according to Nimchua et al. (2008) with slight modifications. Assays were conducted in 2-mL tubes containing 10 mg of PET (ground granules, Sigma-Aldrich), 1 mL of Tris-HCl (20 mM, pH 7.5), and 3 μg of the enzyme Sub1. In the first experiment, the effects of Triton on Sub1 performance were tested. Tubes, with or without Triton X-100 (0.5%), were incubated at 37°C and the concentrations of TA released in the incubation media were recorded after 10 and 15 d. Blank assays, in which the enzyme Sub1 was omitted, were used as controls. Tubes were then centrifuged (1 min) and 50 μL of the collected supernatant was added to 350 μL of Tris-HCl (20 mM, pH 7.5) into quartz cuvettes. Absorbance at 240 nm was measured to assess TA concentrations (using a standard curve). In another experiment, the stability of Sub1, using PET as a substrate, was assessed at 37 and 50°C over a 20-d period. The incubation medium contained Triton X-100 (0.5%) and the concentration of TA released in the reaction mix was measured every 5 d, as described above.
Results
Heterologous production of Sub1
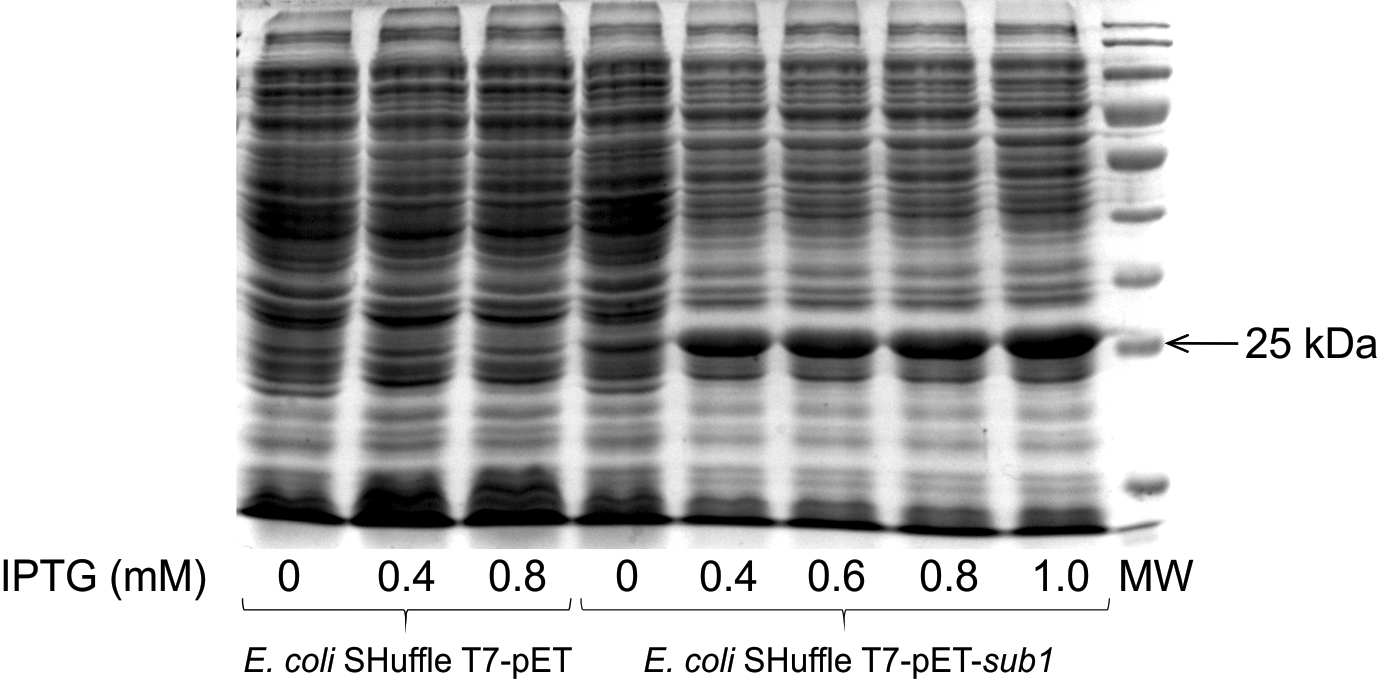
The S. scabies sub1 gene was cloned into a pET expression vector (data not shown) and expressed in E. coli strain SHuffle T7. The cytoplasmic extract of E. coli SHuffle T7-pET-sub1 was characterized by a thick and dense band of approximatively 25 kDa, which was absent from the protein profile of E. coli SHuffle T7-pET (Fig. 1). In E. coli SHuffle T7-pET-sub1, Sub1 was produced even in the absence of IPTG, while thicker protein bands were observed when sub1 gene expression was induced by this compound (Fig. 1).
The cytoplasmic fraction from cultures of E. coli Shuffle T7-pET-sub1 showed, in the absence of the induction with IPTG, that esterase activity on the C4 substrate (58.8 μmol mL–1) was significantly higher than that in control E. coli SHuffle T7-pET (1.6 μmol mL–1) after 30 min of the incubation (Fig. 2). The esterase activity of E. coli SHuffle T7-pET-sub1 was inducible with IPTG and was quickly observable in the reaction mix. After 5 min of the incubation, the highest activity reached 32.2 μmol mL–1 when 0.8 mM IPTG was added to the culture, while activity was 6.3 μmol mL–1 in the absence of IPTG (Fig. 2).
Purification of the recombinant protein His-Sub1
An affinity column Ni-NT (cOmplete His-Tag) was used to purify the recombinant protein Sub1 from the cytoplasmic fraction of E. coli SHuffle T7-pET-sub1. By comparing the migration profile of the cytoplasmic fraction and the flow-through (Fig. 3, lanes 2 and 3, respectively), the band corresponding to the recombinant protein His-Sub1 (25 kDa) was present in the cytoplasmic fraction, while no band was present in the flow-through fraction, indicating that the Sub1 protein bound the column. The presence of imidazole in the elution buffer allows the elution of His-tag proteins from the column. At imidazole concentrations ranging between 4 and 10 mM, the elution buffer released the majority of the contaminant proteins, while a low quantity of Sub1 was released with 10 mM imidazole (lane 7, Fig. 3). A detectable quantity of Sub1 was eluted with no contaminant proteins when 50 mM imidazole buffer was used (lane 8, Fig. 3). However, at 200 mM imidazole, no recombinant protein was detected (lane 9, Fig. 3). A mass spectrometry analysis confirmed that the purified protein was Sub1 (data not shown). Purification efficacy was estimated by comparing the esterase activities (with 0.4 mM p-nitrophenol butyrate as the substrate) of the cytoplasmic fraction and the purified protein. The esterase activity of Sub1 was 52-fold higher in the purified extract (23.0 U mL–1) than in the cytoplasmic crude extract (0.4 U mL–1).

The esterase activity of Sub1 on the p-nitrophenyl esters of varying carbon chain lengths was assessed in the presence and absence of Triton X-100. Independent of the presence of Triton X-100, Sub1 was more active on p-nitrophenyl butyrate (C4) and p-nitrophenyl octanoate (C8) than on p-nitrophenyl esters with longer carbon chains (C10 and C12, Fig. 4). The presence of Triton X-100 increased the esterase activity of the enzyme on all of these substrates (Fig. 4). The highest esterase activity (14.6 U nmol–1 or 616 U mg–1 Sub1) was obtained on the C4 substrate in the presence of Triton X-100. Thus, Vmax and Km values for esterase Sub1 were calculated using the latter substrate. As shown in Fig. 5, the initial velocity (V0) of the hydrolysis reaction increased when higher concentrations of the substrate p-nitrophenol butyrate (p-NPB) were used. According to the Michaelis-Menten equation, Vmax was 55.8±2.0 U nmol–1 Sub1 (2,361±84.5 U mg–1 Sub1) and Km was 0.57±0.04 mM p-NPB.

Sub1 was shown to hydrolyze suberin and cutin, releasing 1.22±0.06 and 2.65±0.18 nmol of fatty acids (palmitic acid equivalent) μg–1 Sub1 from these polymers, respectively, after 20 d of the incubation. The ability of Sub1 to degrade cutin and suberin appeared to be stable over the experimental time course at room temperature because the amounts of fatty acids released in the incubation medium correlated with time (P<0.0001; Fig. 6).

On the other hand, the release of TA showed that the Sub1 esterase also had the ability to hydrolyze the synthetic substrate PET. The addition of Triton X-100 to the reaction mix enhanced the hydrolysis of PET by ca. 2.6-fold (P<0.0001, t-test; Fig. 7A) after 10 and 15 d of the incubation. The esterase activity of Sub1 on PET, in the presence of Triton X-100, increased over the incubation time because the amount of TA released during the degradation of PET correlated with the incubation time (P<0.0001; Fig. 7B). The enzyme Sub1 showed high stability at 37°C over the test period (20 d) because the concentration of TA released in the incubation medium linearly correlated with time (r2=0.9874). However, the stability of Sub1 at 50°C fit a non-linear curve (r2=0.9667).
Discussion
S. scabies may colonize potato tuber surfaces and is able to directly penetrate potato cells (Loria et al., 2003). Previous studies proposed that its entry into potato tuber tissues may be facilitated by the production of esterases that degrade suberin present in the potato periderm (McQueen and Schottel, 1987; Beauséjour et al., 1999; Komeil et al., 2013). Although suberin degradation has not yet been examined in detail, some fungal cutinases exhibit activity towards suberin (Kontkanen et al., 2009). The protein Sub1 is part of the S. scabies secretome when this bacterium is grown in the presence of suberin (Beaulieu et al., 2016) and the sub1 gene is induced in the presence of suberin (Komeil et al., 2013). This study predicted that Sub1 was a cutinase due to its high sequence homology with other cutinases of fungal origin. The present results confirm this prediction because Sub1 exhibited the ability to hydrolyze both cutin and suberin.
In the present study, the heterologous production of the S. scabies Sub1 protein was successfully achieved in E. coli. Other studies also reported the heterologous expression of bacterial esterases in E. coli (Chen et al., 2008; Su et al., 2013; Ribitsch et al., 2015). The molecular weight of His-tagged Sub1 was estimated herein to be 25 kDa, which is consistent with the predicted molecular weight of mature Sub1 (18.7 kDa) plus the His-tag (4.9 kDa). Therefore, the molecular weight of Sub1 appears to be less than that of most bacterial cutinases, such as Tfu-0882 and Tfu-0883 from Thermobifida fusca (29 kDa; Chen et al., 2008), but is closer to those of fungal plant pathogen cutinases, such as CcCUT1 from C. cinerea (18.8 kDa; Kontkanen et al., 2009) and CutA from Botrytis cinerea (18 kDa; van der Vlugt-Bergmans et al., 1997).
As reported with other cutinases, purified Sub1 also has the ability to hydrolyze p-nitrophenyl esters. Sub1 was more active on p-nitrophenyl esters with short carbon chains, i.e.
p-nitrophenyl butyrate (p-NPB, C4) and p-nitrophenyl octanoate (C8), than on those with longer carbon chains (C10 and C12). Other microbial cutinases have also been reported to be more active on p-nitrophenyl esters harboring short fatty acid chains (Purdy and Kolattukudy, 1973; Kontkanen et al., 2009). Using p-nitrophenyl butyrate as a substrate, the activity of Sub1 followed a typical Michaelis-Menten curve. The Sub1 enzyme showed affinity towards this substrate (Km=5.7 10–4 M), similar to two cutinases of Fusarium solani pv. pisi with Km of 3.5 10–4 M and of 7.5 10–4 M, respectively (Kolattukudy et al., 1981). Although streptomycetes such as S. scabies and filamentous fungi belong to different kingdoms, they exhibit similar lifestyles and often share the same ecological niches (Wösten and Willey, 2000). Their mycelia colonize various organic polymers and produce large amounts of extracellular enzymes to retrieve nutrients from these substrates. Therefore, similarities between Sub1 and fungal cutinases may reflect an adaptation to a similar lifestyle. Other Streptomyces extracellular enzymes, e.g. chitosanases from the GH75 family, are encoded by genes that are also mainly represented in fungal and actinobacterial genomes (Lacombe-Harvey et al., 2018).
Sub1 has high similarity, at the amino acid level, to the cutinase CUT1 from F. solani, which is able to degrade suberin. Similar to CUT1, the present results indicated that Sub1 exhibits hydrolysis activity on suberin. However, Sub1 and CUT1 showed higher activity on cutin than on suberin, even though S. scabies and F. solani both infect and colonize potato tubers and are, thus, frequently in contact with potato suberin (Fiers et al., 2012). This finding indicates that the presence of aromatic compounds in suberin also contributes to its recalcitrant nature (Faber, 1979). However, difficulties are associated with comparing the efficacy of Sub1 with fungal cutinases because the methods used to monitor cutinase activity differ between studies (van der Vlugt-Bergmans et al., 1997; Kontkanen et al., 2009; Chen et al., 2013; the present results). To the best of our knowledge, Sub1 represents the first bacterial cutinase for which activity towards suberin has been demonstrated. Based on the ecological niche of this pathogen, in which potato tubers, but not the aerial part of the plant, are infected, suberin most likely represents an important substrate for Sub1 in the environment and, consequently, Sub1 may be designated as a suberinase. While the ability to degrade cutin has been shown to be important for the pathogenicity of various fungal plant pathogens (Feng et al., 2011; Wang et al., 2017), there is no evidence to show that the ability to degrade suberin represents an asset in the infection process of plant pathogens. The Sub1 protein does not appear to be an essential pathogenicity factor for S. scabies because the sub1 gene has not been detected in other common scab-inducing Streptomyces species, such as S. acidiscabies (Komeil et al., 2013), and its primary benefit may involve the degradation of refractory polymers. However, Sub1 may confer an advantage to S. scabies over other common scab-inducing species by facilitating direct penetration, tuber colonization, and persistence in potato tuber debris.
While Sub1 is conserved in only a few streptomycetes, it presents high homology not only with some fungal cutinases, but also with cutinase-like enzymes from animal pathogenic mycobacteria (Komeil et al., 2013). These bacteria do not encounter cutin or suberin in their environment; however, cutinases were identified as multifunctional enzymes that act on phospholipids, polysorbates, triacylglycerols, and triolein (Schué et al., 2010; Monu and Meena, 2016). As multifunctional enzymes, cutinases have a number of applications in industry (Carvalho et al., 1998). In the present study, the effects of Sub1 on PET, a synthetic polyester that is widely used in the production of textiles, were tested. Sub1 was shown to degrade PET because the quantity of terephthalic acid released from the synthetic polymer depended on the enzyme concentration and increased over the incubation time. The enzyme Sub1 maintained its activity at 37°C for at least 20 d, showing that it is stable, as has been demonstrated for other cutinases (Dutta et al., 2009). Due to their functional properties, cutinases are considered to be a link between esterases and lipases (Chahinian et al., 2002). The addition of a non-ionic surfactant, such as Triton, into a reaction mix generally promotes the activity of lipases, but does not affect the activity of most cutinases. For example, the presence of Triton in the reaction mix increased the hydrolysis of polyester bis-(benzoyloxyethyl) terephthalate (3PET) by a lipase secreted by Thermomyces lanuginosus, whereas it did not exert any effect when the same substrate was exposed to cutinases secreted by T. fusca and F. solani (Eberl et al., 2009). Triton increases the activity of lipases by promoting the opening of a peptide lid located over the active site of the enzyme; such a lid is not present in cutinases (Eberl et al., 2009). As observed with most cutinases, the addition of Triton X-100 did not affect cutin hydrolysis by Sub1 (data not shown). Nevertheless, the hydrolysis of p-nitrophenyl esters and of PET by Sub1 was enhanced in the presence of Triton.
The present study established that the sub1 gene of S. scabies encodes a protein acting as a suberinase. The versatility of Sub1 may also be considered for adoption in industrial applications. A cutinase-like enzyme has recently attracted global public attention. This enzyme, originally characterized in the bacterium Ideonella sakaiensis 201-F6, is able to degrade PET and has been reclassified as a PETase (Yoshida et al., 2016). Thereafter, Austin et al. (2018) introduced modifications to the binding cleft of this enzyme and produced an engineered PETase with better plastic-degrading properties than the native enzyme. The current need for enzymes with the ability to degrade refractory polymers of anthropic origin makes further studies on the capabilities of Sub1 very relevant.
Acknowledgements
The authors thank Peter Moffett for the critical review of the manuscript. This work was funded by the Natural Sciences and Engineering Research Council of Canada (grant number 018602).
References
- Austin, H.P., Allen, M.D., Donohoe, B.S., Rorrer, N.A., Kearns, F.L., Silveira, R.L., et al. (2018) Characterization and engineering of a plastic-degrading aromatic polyesterase. Proc Natl Acad Sci U S A
115: E4350–E4357.
- Beaulieu, C., Sidibé, A., Jabloune, R., Simao-Beaunoir, A.-M., Lerat, S., Monga, E., and Bernards, M.A. (2016) Physical, chemical and proteomic evidence of potato suberin degradation by the plant pathogenic bacterium Streptomyces scabiei. Microbes Environ
31: 427–434.
- Beauséjour, J., Goyer, C., Vachon, J., and Beaulieu, C. (1999) Production of thaxtomin A by Streptomyces scabies strains in plant extract containing media. Can J Microbiol
45: 764–768.
- Beisson, F., Li-Beisson, Y., and Pollard, M. (2012) Solving the puzzles of cutin and suberin polymer biosynthesis. Curr Opin Plant Biol
15: 329–337.
- Bernards, M.A., and Lewis, N.G. (1998) The macromolecular aromatic domain in suberized tissue: a changing paradigm. Phytochemistry
47: 915–933.
- Bernards, M.A. (2002) Demystifying suberin. Can J Bot
80: 227–240.
- Bradford, M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem
72: 248–254.
- Carvalho, C.M.L., Aires-Barros, M.R., and Cabral, J.M.S. (1998) Cutinase structure, function and biocatalytic applications. Electron J Biotechnol
1: 160–173.
- Carvalho, C.M.L., Aires-Barros, M.R., and Cabral, J.M.S. (1999) Cutinase: from molecular level to bioprocess development. Biotechnol Bioeng
66: 17–34.
- Chahinian, H., Nini, L., Boitard, E., Dubès, J.-P., Comeau, L.-C., and Sarda, L. (2002) Distinction between esterases and lipases: a kinetic study with vinyl esters and TAG. Lipids
37: 653–662.
- Chen, S., Tong, X., Woodard, R.W., Du, G., Wu, J., and Chen, J. (2008) Identification and characterization of bacterial cutinase. J Biol Chem
283: 25854–25862.
- Chen, S., Su, L., Chen, J., and Wu, J. (2013) Cutinase: characteristics, preparation, and application. Biotechnol Adv
31: 1754–1767.
- Dutta, K., Sen, S., and Veeranki, V.D. (2009) Production, characterization and applications of microbial cutinases. Process Biochem
44: 127–134.
- Eberl, A., Heumann, S., Brückner, T., Araujo, R., Cavaco-Paulo, A., Kaufmann, F., et al. (2009) Enzymatic surface hydrolysis of poly(ethylene terephthalate) and bis(benzoyloxyethyl) terephthalate by lipase and cutinase in the presence of surface active molecules. J Biotechnol
143: 207–212.
- Faber, M.D. (1979) Microbial degradation of recalcitrant compounds and synthetic aromatic polymers. Enzyme Microb Technol
1: 226–232.
- Feng, J., Hwang, R., Hwang, S.-F., Gaudet, D., and Strelkov, S.E. (2011) Molecular characterization of a Stagonospora nodorum lipase gene LIP1. Plant Pathol
60: 698–708.
- Fiers, M., Edel-Hermann, V., Chatot, C., Le Hingrat, Y., Alabouvette, C., and Steinberg, C. (2012) Potato soil-borne diseases. A review. Agron Sustainable Dev
32: 93–132.
- Hill, J., and Lazarovits, G. (2005) A mail survey of growers to estimate potato common scab prevalence and economic loss in Canada. Can J Plant Pathol
27: 46–52.
- Khatri, B.B., Tegg, R.S., Brown, P.H., and Wilson, C.R. (2011) Temporal association of potato tuber development with susceptibility to common scab and Streptomyces scabiei-induced responses in the potato periderm. Plant Pathol
60: 776–786.
- Kolattukudy, P.E., and Agrawal, V.P. (1974) Structure and composition of aliphatic constituents of potato tuber skin (suberin). Lipids
9: 682–691.
- Kolattukudy, P., Purdy, R., and Maiti, I. (1981) Cutinases from fungi and pollen. Methods Enzymol
71: 652–664.
- Komeil, D., Simao-Beaunoir, A.-M., and Beaulieu, C. (2013) Detection of potential suberinase-encoding genes in Streptomyces scabiei strains and other actinobacteria. Can J Microbiol
59: 294–303.
- Komeil, D., Padilla-Reynaud, R., Lerat, S., Simao-Beaunoir, A.-M., and Beaulieu, C. (2014) Comparative secretome analysis of Streptomyces scabiei during growth in the presence or absence of potato suberin. Proteome Sci
12: 35.
- Kontkanen, H., Westerholm-Parvinen, A., Saloheimo, M., Bailey, M., Rättö, M., Mattila, I., et al. (2009) Novel Coprinopsis cinerea polyesterase that hydrolyzes cutin and suberin. Appl Environ Microbiol
75: 2148–2157.
- Lacombe-Harvey, M.-È., Brzezinski, R., and Beaulieu, C. (2018) Chitinolytic functions in actinobacteria : ecology, enzymes, and evolution. Appl Microbiol Biotechnol
102: 7219–7230.
- Lauzier, A., Simao-Beaunoir, A.-M., Bourassa, S., Poirier, G.G., Talbot, B., and Beaulieu, C. (2008) Effect of potato suberin on Streptomyces scabies proteome. Mol Plant Pathol
9: 753–762.
- Liu, Z., Gosser, Y., Baker, P.J., Ravee, Y., Lu, Z., Alemu, G., et al. (2009) Structural and functional studies of Aspergillus oryzae cutinase: enhanced thermostability and hydrolytic activity of synthetic ester and polyester degradation. J Am Chem Soc
131: 15711–15716.
- Loria, R., Coombs, J., Yoshida, M., Kers, J.A., and Bukhalid, R.A. (2003) A paucity of bacterial root diseases : Streptomyces succeeds where others fail. Physiol Mol Plant Pathol
62: 65–72.
- Martinez, C., Nicolas, A., van Tilbeurgh, H., Egloff, M.P., Cudrey, C., Verger, R., and Cambillau, C. (1994) Cutinase, a lipolytic enzyme with a preformed oxyanion hole. Biochemistry
33: 83–89.
- McQueen, D.A.R., and Schottel, J.L. (1987) Purification and characterization of a novel extracellular esterase from pathogenic Streptomyces scabies that is inducible by zinc. J Bacteriol
169: 1967–1971.
- Monu, and Meena, L.S. (2016) Roles of triolein and lipolytic protein in the pathogenesis and survival of Mycobacterium tuberculosis: a novel therapeutic approach. Appl Biochem Biotechnol
178: 1377–1389.
- Murphy, C.A., Cameron, J.A., Huang, S.J., and Vinopal, R.T. (1996)
Fusarium polycaprolactone depolymerase is cutinase. Appl Environ Microbiol
62: 456–460.
- Nimchua, T., Eveleigh, D.E., Sangwatanaroj, U., and Punnapayak, H. (2008) Screening of tropical fungi producing polyethylene terephthalate-hydrolyzing enzyme for fabric modification. J Ind Microbiol Biotechnol
35: 843–850.
- Purdy, R.E., and Kolattukudy, P.E. (1973) Depolymerization of a hydroxy fatty acid biopolymer, cutin, by an extracellular enzyme from Fusarium solani f. pisi: isolation and some properties of the enzyme. Arch Biochem Biophys
159: 61–69.
- Ribitsch, D., Acero, E.H., Przylucka, A., Zitzenbacher, S., Marold, A., Gamerith, C., et al. (2015) Enhanced cutinase-catalyzed hydrolysis of polyethylene terephthalate by covalent fusion to hydrophobins. Appl Environ Microbiol
81: 3586–3592.
- Sambrook, J.F., and Russell, D.W. (2001) Molecular Cloning: A Laboratory Manual, 3rd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Schué, M., Maurin, D., Dhouib, R., Bakala N’Goma, J.-C., Delorme, V., Lambeau, G., et al. (2010) Two cutinase-like proteins secreted by Mycobacterium tuberculosis show very different lipolytic activities reflecting their physiological function. FASEB J
24: 1631–2136.
- Su, L., Woodard, R.W., Chen, J., and Wu, J. (2013) Extracellular location of Thermobifida fusca cutinase expressed in Escherichia coli BL21(DE3) without mediation of a signal peptide. Appl Environ Microbiol
79: 4192–4198.
- Tyner, D.N., Hocart, M.J., Lennard, J.H., and Graham, D.C. (1997) Periderm and lenticel characterization in relation to potato cultivar, soil moisture and tuber maturity. Potato Res
40: 181–190.
- van der Vlugt-Bergmans, C.J., Wagemakers, C.A., and van Kan, J.A. (1997) Cloning and expression of the cutinase A gene of Botrytis cinerea. Mol Plant Microbe Interact
10: 21–29.
- Vertommen, M.A.M.E., Nierstrasz, V.A., van der Veer, M., and Warmoeskerken, M.M.C.G. (2005) Enzymatic surface modification of poly(ethylene terephthalate). J Biotechnol
120: 376–386.
- Walton, T.J., and Kolattukudy, P.E. (1972) Determination of the structures of cutin monomers by a novel depolymerization procedure and combined gas chromatography and mass spectrometry. Biochemistry
11: 1885–1897.
- Wang, Y., Chen, J., Li, D.W., Zheng, L., and Huang, J. (2017)
CglCUT1 gene required for cutinase activity and pathogenicity of Colletotrichum gloeosporioides causing anthracnose of Camellia oleifera. Eur J Plant Pathol
147: 103–114.
- Wösten, H.A.B., and Willey, J.M. (2000) Surface-active proteins enable microbial aerial hyphae to grow into the air. Microbiology
146: 767–773.
- Yoshida, S., Hiraga, K., Takehana, T., Taniguchi, I., Yamaji, H., Maeda, Y., et al. (2016) A bacterium that degrades and assimilates poly(ethylene terephthalate). Science
351: 1196–1199.


