Abstract
Uncultivated members of Candidatus Patescibacteria are commonly found in activated sludge treating sewage and are widely distributed in wastewater treatment plants in different regions and countries. However, the phylogenetic diversity of Ca. Patescibacteria is difficult to examine because of their low relative abundance in the environment. Since Ca. Patescibacteria members have small cell sizes, we herein collected small microorganisms from activated sludge using a filtration-based size-fractionation approach (i.e., 0.45–0.22 μm and 0.22–0.1 μm fractions). Fractionated samples were characterized using 16S rRNA gene amplicon and shotgun metagenomic sequence analyses. The amplicon analysis revealed that the relative abundance of Ca. Patescibacteria increased to 73.5% and 52.5% in the 0.45–0.22 μm and 0.22–0.1 μm fraction samples, respectively, from 5.8% in the unfractionated sample. The members recovered from the two size-fractionated samples included Ca. Saccharimonadia, Ca. Gracilibacteria, Ca. Paceibacteria, Ca. Microgenomatia, class-level uncultured lineage ABY1, Ca. Berkelbacteria, WS6 (Ca. Dojkabacteria), and WWE3, with Ca. Saccharimonadia being predominant in both fraction samples. The number of operational taxonomic units belonging to Ca. Patescibacteria was approximately 6-fold higher in the size-fractionated samples than in the unfractionated sample. The shotgun metagenomic analysis of the 0.45–0.22 μm fractioned sample enabled the reconstruction of 24 high-quality patescibacterial bins. The bins obtained were classified into diverse clades at the family and genus levels, some of which were rarely detected in previous activated sludge studies. Collectively, the present results suggest that the overall diversity of Ca. Patescibacteria inhabiting activated sludge is higher than previously expected.
Activated sludge processes have been applied to the treatment of various types of wastewater, including municipal and industrial wastewater. Microorganisms in activated sludge are key players in the removal of various pollutants (e.g., organic matter, nitrogen, and phosphorus compounds). The microbial composition of activated sludge is affected by the type of wastewater, microorganisms in influent wastewater, the location of treatment plants, treatment processes, and operating conditions (e.g., solid retention time) (Gonzalez-Martinez et al., 2016; Xu et al., 2018). Therefore, further knowledge of microbial phylogenetic diversity and abundance in various wastewater treatment processes is needed to obtain a more detailed understanding of activated sludge ecosystems. Although the morphology, diversity, physiology, and distribution of microorganisms involved in wastewater treatment are being documented and provided in databases, such as MiDAS (Nierychlo et al., 2020), the functional roles of most microorganisms in activated sludge systems remain unclear because of the highly complex ecosystem as well as the presence of yet-to-be-cultured microorganisms (Chouari et al., 2010; Sekiguchi et al., 2015; Kindaichi et al., 2016; Wang et al., 2018).
Microorganisms belonging to Candidatus Patescibacteria (also known as candidate phyla radiation) are often found in wastewater treatment systems (Hu et al., 2012; Zhang et al., 2012; Ju and Zhang, 2015; Kindaichi et al., 2016; Chin et al., 2017; Li et al., 2020; Hosokawa et al., 2021; Martínez-Campos et al., 2021; Singleton et al., 2021). They also exist in activated sludge processes treating sewage, and major classes are Candidatus Saccharimonadia (TM7), Candidatus Paceibacteria (OD1, also known as Candidatus Parcubacteria), and Candidatus Gracilibacteria (GN02/BD1-5). We hereafter omit “Candidatus” to describe the patescibacterial phylogenetic groups for brevity. Patescibacteria is a taxonomic group with few culture representatives (e.g., TM7x [He et al., 2015]), and culture-independent approaches (e.g., microautoradiography and fluorescence in situ hybridization [MAR-FISH] as well as metagenomics) have been adapted to elucidate their functions and roles in wastewater treatment processes. Saccharimonadia contributes to the degradation of sugar compounds in municipal wastewater treatment processes (Nielsen et al., 2009; Kindaichi et al., 2016). Saccharimonadia have also been suggested to be involved in ammoniacal nitrogen removal (Remmas et al., 2017). Paceibacteria, Microgenomatia, and Gracilibacteria adapt to an anaerobic fermentation-based lifestyle and may play a role in hydrogen production and/or sulfur cycling (Wrighton et al., 2012). In addition, Patescibacteria in an anammox reactor are expected to play roles in degrading the metabolic products of anammox bacteria and providing lactate and formate to other bacteria in order to maintain the anammox ecosystem (Hosokawa et al., 2021).
Patescibacteria found in sludge treating wastewater are phylogenetically distinct among the samples analyzed; e.g., Saccharimonadia found in sewage treatment plants in different countries and regions are not closely related (Kindaichi et al., 2016). This implies that Patescibacteria involved in wastewater treatment are diverse, and their roles in activated sludge ecosystems may vary under different wastewater treatment operations. To obtain a more detailed understanding of Patescibacteria in activated sludge, a selective in-depth analysis is crucial; however, it is hampered by the relatively low abundance of Patescibacteria in the complex microbial communities of activated sludge.
Patescibacteria is characterized by a small cell size (median diameter of 0.2 μm, referred to as ultramicrobacteria) (Beam et al., 2020). Studies on Patescibacteria in natural environments, such as seawater (Tully et al., 2018), groundwater (Luef et al., 2015; Ludington et al., 2017; Probst et al., 2017; Tian et al., 2020; Chaudhari et al., 2021), and freshwater lakes (Vigneron et al., 2019) often involve performing the enrichment of small microorganisms by size fractionation as a pre-treatment process. This approach may be effective for the recovery of Patescibacteria from activated sludge. The present study attempted to enrich small microorganisms in sludge samples using a filtration-based size-fractionation approach and to elucidate the phylogenetic diversities of Patescibacteria in size-fractionated samples using 16S rRNA gene amplicon and metagenomic analyses.
Materials and Methods
Sample collection and size fractionation
An activated sludge sample (designated as “unfractionated sludge”) was collected from the reaction tank of a wastewater treatment plant (WWTP) employing a conventional activated sludge process in Miyagi, Japan. The activated sludge process was operated at a solid retention time of 5.3 d and hydraulic retention time of 5.7 h. Mixed liquor suspended solid and dissolved oxygen concentrations were 958 and 1.25 mg L–1, respectively, and pH was 6.8 in the reaction tank at the time of sampling. Quality parameters for influent water to the reaction tank and effluent water from the final sedimentation tank were 84 and 6 mg L–1 for biochemical oxygen demand, 26 and 27 mg L–1 for total nitrogen, and 3.6 and 0.7 mg L–1 for total phosphorus, respectively (average values of the sampling month). Regarding size fractionation, the activated sludge sample was centrifuged (7,000×g at 4°C for 5 min). The activated sludge supernatant obtained was serially filtered through filters of decreasing pore sizes using a Stericup® filter unit (filter area: 40 cm2, Merck KGaA): 0.45 μm (PVDF membrane, S2HVU02RE), 0.22 μm (PVDF membrane, S2GVU02RE), and 0.1 μm (polyethersulfone membrane, S2VPU02RE). The 0.22- and 0.1-μm filters were used as fractionated samples (designated as “0.45–0.22 μm fraction” and “0.22–0.1 μm fraction”, respectively). Samples were stored at –20°C until used.
Fluorescence in situ hybridization (FISH)
The filtrate of the 0.45-μm filter was fixed with 2% paraformaldehyde at 4°C for 24 h. Subsequently, 30–50 mL of the fixed sample was filtered through a polycarbonate membrane filter with a pore size of 0.2 μm (diameter of 25 mm; ADVANTEC). After drying, filters were dipped in 0.1% low-melting-point agarose (Invitrogen) in ultrapure water (w/v) for embedding (Pernthaler et al., 2002). The filters were dried at room temperature and cut into 16 pieces. The TM7567 (5′-CCT ACG CAA CTC TTT ACG CC-3′) and TM7305 (5′-GTC CCA GTC TGG CTG ATC-3′) probes (Hugenholtz et al., 2001) were used to detect Saccharimonadia. Both probes were labeled with Alexa Fluor 555 at the 5′-end. The TM7567 probe matched most of the 16S rRNA gene sequences of saccharimonadial operational taxonomic units (OTUs) obtained using the amplicon analysis and bins obtained using the metagenomic analysis (Fig. S1). Regarding hybridization, filters were incubated in hybridization buffer (20 mM Tris-HCl [pH 7.5], 900 mM NaCl, 1% blocking reagent [w/v] [Roche], and 0.01% sodium dodecyl sulfate [SDS, w/v]) containing 0.5 μM of the probe at 46°C for 3 h. A specific formamide concentration was used for each probe, i.e., 30% for TM7567 and 40% for TM7305. After the incubation, the filters were immersed in washing buffer (20 mM Tris-HCl [pH 7.5], 0.01% SDS [w/v], 5 mM EDTA, 103 mM NaCl for TM7567, and 46 mM NaCl for TM7305) at 48°C for 15 min. The filters were dried and stained with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI, 1 μg mL–1; Merck KGaA). Microscopic observations were performed using Axio Imager 2 (Carl Zeiss) equipped with AxioCam HRm (Carl Zeiss).
DNA extraction
DNA extraction from the unfractionated sludge sample was performed using the ISOIL for Beads Beating Kit (NIPPON GENE) by applying 0.1 g wet sample. Half of the filter (approximately 20 cm2) was used for fractionated samples. The filters were finely chopped into small pieces, immersed in 1.055 mL of proteinase K solution (0.1 mg mL–1 in 50 mM Tris-HCl [pH 8.0], 100 mM CaCl2, and 0.5% SDS), and incubated at 37°C for 3 h. Six hundred microliters of the solution was subjected to the ISOIL for Beads Beating Kit (DNA purification process only). DNA was eluted in 100 μL TE buffer (10 mM Tris-HCl and 1 mM EDTA [pH 8.0]). DNA concentrations were measured using a Qubit® 3 Fluorometer (Thermo Fisher Scientific) with the Qubit® dsDNA HS Assay Kit (Thermo Fisher Scientific). The DNA yield of fractionated samples was calculated as follows: DNA concentrations were multiplied by the elution volume and divided by the number of filters used for DNA extraction.
Amplicon analysis targeting the 16S rRNA gene
Amplicon sequencing was performed to target the V3–V4 region of the 16S rRNA gene (Ni et al., 2020). Primers without overhang sequences were used for the initial polymerase chain reaction (PCR) to minimize PCR bias (Berry et al., 2011). The primer sets of 341F (5′-CCT AYG GGR BGC ASC AG-3′) and 806R-mix (a mixture of 806R [5′-GGA CTA CHV GGG THT CTA AT-3′] and 806R-P [5′-GGA CTA CCA GGG TAT CTA AG-3′] at a ratio of 30:1) were used (Matsubayashi et al., 2017). The first round of PCR was performed using TaKaRa TaqTM HS Low DNA (Takara Bio) and consisted of 25 amplification cycles (94°C for 5 s, 50°C for 5 s, and 68°C for 10 s), followed by a final extension step at 68°C for 7 min. Forty amplification cycles were performed for the fractionated samples. PCR products were purified using Agencourt AMPure® XP (Beckman Coulter). Primers with overhang sequences were used for the second round of PCR. The reaction was performed using TaKaRa Ex Taq® Hot Start Version (Takara Bio) and consisted of a denaturing step at 94°C for 2 min, followed by five amplification cycles (94°C for 30 s, 50°C for 30 s, and 72°C for 30 s), and a final extension step at 72°C for 5 min. Purification was performed as previously described. The primers in Nextera® XT Index Kit v2 Set A (Illumina) were used for the third round of PCR. The third round of PCR was performed using TaKaRa Ex Taq® Hot Start Version, consisting of a denaturing step at 94°C for 2 min, followed by eight amplification cycles (94°C for 30 s, 55°C for 30 s, and 72°C for 30 s), and a final extension step at 72°C for 5 min. PCR products were verified for specific amplification using an Agilent Technology 2100 Bioanalyzer and Agilent DNA7500 Kit (Agilent Technology) and purified using Agencourt AMPure® XP. The DNA concentration of the purified product was measured using a Qubit® 3 Fluorometer with the Qubit® dsDNA HS Assay Kit and adjusted to 4 nM by diluting it with 10 mM Tris-HCl (pH 8.0). Sequencing of the products was conducted using an Illumina MiSeq sequencer (Illumina) with the MiSeq Reagent Kit v3 600-cycles (Illumina), following the manufacturer’s instructions.
Sequence data were initially subjected to a quality check (Trimmomatic ver. 0.39, SLIDINGWINDOW:6:20 MINLEN:200) (Bolger et al., 2014). Passed sequence data were paired and checked for quality (read lengths: 300–500 bps, quality score: higher than 25) and chimeras (usearch61) using QIIMETM ver. 1.8.0 software (Caporaso et al., 2010). Single reads were removed using VSEARCH (Rognes et al., 2016). OTUs were generated using a 97% sequence identity threshold. The SILVA 132 database (Quast et al., 2013) was used for assignments. In this database, Paceibacteria are referred to as Parcubacteria. In the present study, we described Parcubacteria as Paceibacteria. OTUs assigned as “None” or “No blast hit” were reanalyzed with NCBI Blast (megablast, https://blast.ncbi.nlm.nih.gov/Blast.cgi) using the representative sequence of each OTU. As a result, approximately 90% of the OTUs assigned as “None” or “No blast hit” failed to match any species. Other OTUs hit viruses or plants with a low sequence identity. Therefore, OTUs assigned as “None” or “No blast hit” were removed from the analysis. The number of sequence reads obtained were 26,078 from the unfractionated sludge sample, 25,035 from the 0.45–0.22 μm fraction sample, and 7,383 from the 0.22–0.1 μm fraction sample. To maintain the same sequencing depth, 7,000 randomly selected sequence reads were used for further data analyses. The Goods coverage was 96.4–98.3%.
Metagenomic analysis
A metagenomic analysis was performed using the 0.45–0.22 μm fraction sample. Prior to library preparation, ethanol precipitation was conducted to concentrate DNA. The Nextera XT DNA Library Preparation Kit (Illumina) was used for library preparation. The prepared library was sequenced using a MiSeq sequencer with the MiSeq Reagent Kit v2 500-cycles (Illumina). Sequencing was performed twice using the same library.
Sequence data were trimmed based on the quality score using Trimmomatic ver. 0.39 (SLIDINGWINDOW:6:15, MINLEN:100 for data from the first sequencing run; SLIDINGWINDOW:6:30, MINLEN:100 for data from the second sequencing run). After co-assembly using the MEGAHIT ver. 1.2.9 (k-min 27, k-max 141, k-step 12) (Li et al., 2015; 2016), contigs below 2,500 bp were removed. Contigs were subsequently binned using the MetaBat2 ver. 2.15 (default settings) (Kang et al., 2019), MaxBin 2 ver. 2.2.7 (markerset 40) (Wu et al., 2014), Vamb ver. 3.0.2 (default setting) (Nissen et al., 2021), and MyCC (MyCC_2017. ova) (Lin and Liao, 2016). All four sets of binned metagenomes were subsequently analyzed using DAS Tool ver. 1.1.2 (default settings) (Sieber et al., 2018). Optimized non-redundant bins were dereplicated using dRep ver. 3.2.0 (Olm et al., 2017) with the following parameter: -comp, 50. Quality checks of the bins were performed using CheckM ver. 1.0.7 with the marker set of cpr_43_markers.hmm for Patescibacteria genomes (Parks et al., 2015). Bins with more than 50% completeness and contamination of 10% or less were selected and annotated using Prokka ver. 1.14.6 (Seemann, 2014). The Genome Taxonomy Database Toolkit (GTDB-Tk) ver. 1.5.1 (r202) (Chaumeil et al., 2019) was used for the phylogenetic classification. The 16S rRNA gene sequences recovered from the bins were phylogenetically classified using the SILVA SSU Ref NR 99 132 database (Quast et al., 2013). The BAM file was obtained using BBtool (bbmap.sh, ver. 38.18, input file1=trimmed forward read.fastq, input file2=trimmed reverse read.fastq, reference file=contig.fasta) and SAM tool ver. 1.4.1 (Li et al., 2009). The median coverage of the bins was calculated using CoverM (-m relative abundance; https://github.com/wwood/CoverM) using the BAM file.
Construction of phylogenetic trees
A phylogenetic tree was constructed based on the 16S rRNA gene sequences of Saccharimonadia, Gracilibacteria, Paceibacteria, Microgenomatia, and ABY1 obtained from amplicon and metagenomic analyses. ARB software (http://www.arb-home.de/) (Ludwig et al., 2004) and the SILVA SSU Ref NR 99 132 database (Quast et al., 2013) were used. The phylogenetic tree was constructed using the neighbor-joining method implemented in ARB by extracting representative sequences of Saccharimonadia, Gracilibacteria, Paceibacteria, Microgenomatia, and ABY1 from the database. The 16S rRNA gene sequences obtained through amplicon (368 sequences) and metagenomic (18 sequences) analyses were added to the constructed tree using the parsimony method.
A genome tree was constructed using the sequences of the obtained bins, Patescibacteria sequences recovered from WWTPs in Germany (Schneider et al., 2021) and Denmark (Singleton et al., 2021), and sequences from GTDB-Tk ver. 1.5.1 (r202). The tree was constructed using IQ-Tree 2 ver. 2.1.4-bet (Minh et al., 2020) by the maximum-likelihood (ML) method (IQ-TREE multicore, <alignmentfile> bb 1000 -m LG+G4+FO+I). The model proposed by He et al. was referred to (He et al., 2021).
Deposition of DNA sequence data
The raw sequence data and sequence data of the metagenomic bins were deposited in the DDBJ Sequence Read Archive database (DRA013859).
Results
Microscopic observation of size-fractionated activated sludge
Microorganisms that passed through a pore size of 0.45 μm and were trapped on the 0.2-μm filter were initially stained with DAPI, and we observed small coccoid cells (Fig. 1A). These results clearly indicated the presence of filterable ultramicrobacteria in activated sludge. Patescibacteria members, the target microorganisms of the present study, have a small cell size (Beam et al., 2020). Therefore, we evaluated the sample by applying FISH to detect Saccharimonadia, a member of Patescibacteria known to exist in activated sludge. Two Saccharimonadia-specific probes, TM7305 and TM7567, were used, and the presence of Saccharimonadia in the size-fractionated sample was confirmed by detecting small coccoid-like cells with the TM7567 probe (Fig. 1B), but not with the TM7305 probe. Candidatus Saccharimonas aalborgensis from the activated sludge sample (Albertsen et al., 2013) and TM7x from the human oral sample (He et al., 2015; Bor et al., 2018) were previously reported to have cell sizes of approximately 0.7 and 0.2–0.3 μm, respectively. Similarly, our FISH experiment revealed that Saccharimonadia in the fractionated sample had a small cell size (expected to be between 0.2 and 0.45 μm in diameter).

Microorganisms in the supernatant after centrifuging the activated sludge sample were fractionated into the 0.45–0.22 μm and 0.22–0.1 μm fractions. The volume of the filtrate that passed through the 0.22- and 0.1-μm filters was approximately 3 L each. The concentrations of extracted DNA were 0.21 ng μL–1 (0.45–0.22 μm fraction sample) and 2.48 ng μL–1 (0.22–0.1 μm fraction sample). These DNA yields were equivalent to 0.04 μg filter–1 (0.45–0.22 μm fraction sample) and 0.50 μg filter–1 (0.22–0.1 μm fraction sample). Similar or higher DNA yields (average 1.19 μg filter–1 [0.04–2.84 μg filter–1, minimum to maximum] for 0.45–0.22 μm fraction samples [n=6] and average 3.76 μg filter–1 [0.22–15.10 μg filter–1] for 0.22–0.1 μm fraction samples [n=6]) were obtained from other activated sludge samples (these samples were not analyzed in the present study).
Microbial community structures of fractionated activated sludge samples
The microbial community structures of the unfractionated sludge and the fractionated samples (i.e., the 0.45–0.22 μm and 0.22–0.1 μm fractions) are shown in Table S1, and patescibacterial communities are highlighted in Fig. 2. The relative abundance of Patescibacteria was 73.5% for the 0.45–0.22 μm fraction sample and 52.5% for the 0.22–0.1 μm fraction sample. These values were higher than that of the unfractionated sludge sample of 5.8%. In the unfractionated sludge sample, five phylogenetic groups were detected in Patescibacteria: Saccharimonadia, Gracilibacteria, Paceibacteria, Microgenomatia, and ABY1, among which Saccharimonadia was dominant. In addition to these phylogenetic groups, Berkelbacteria, WS6 (Dojkabacteria), and WWE3 were detected in the fractionated samples. Saccharimonadia, Gracilibacteria, Paceibacteria, Microgenomatia, and ABY1 accounted for ≥1% of the relative abundance in the fractionated samples. Saccharimonadia was the dominant Patescibacteria, accounting for 59.7–68.1% (Fig. S2). Gracilibacteria showed a higher relative abundance in the 0.45–0.22 μm fraction sample than in the 0.22–0.1 μm fraction sample, whereas Paceibacteria, Microgenomatia, and ABY1 showed a higher relative abundance in the 0.22–0.1 μm fraction sample than in the 0.45–0.22 μm fraction sample. These results suggest that the degree of enrichment of Patescibacteria was influenced by the pore size of the filter used for size fractionation.
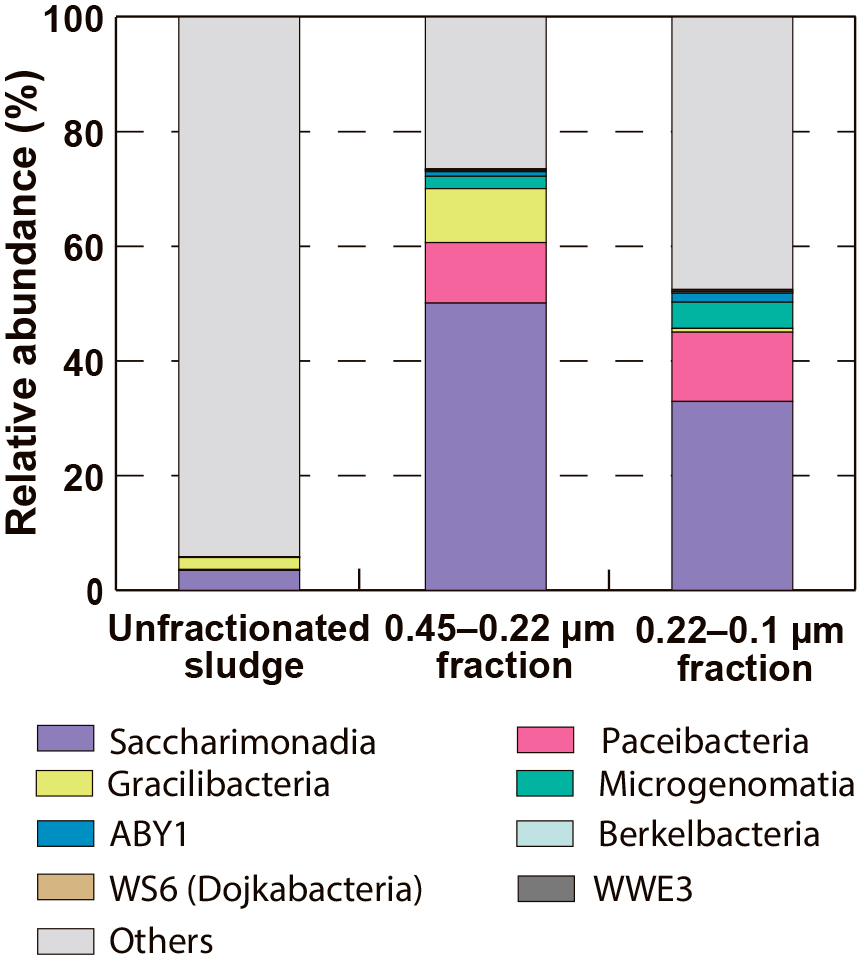
In addition to Patescibacteria, members of Mollicutes (Brown et al., 2007), Bdellovibrio (Rendulic et al., 2004), Polynucleobacter (Hahn, 2003), and Candidatus Woesearchaeia (Castelle et al., 2015) were detected in the fractionated samples. Some of these microorganisms have already been reported as ultramicrobacterial members that pass through 0.45- or 0.22-μm micropore filters (Nakai, 2020).
Diversity of Patescibacteria in activated sludge
The numbers of OTUs and diversity indices of Patescibacteria were higher in the fractionated samples than in the unfractionated sludge sample. Table 1 shows the number of OTUs of Saccharimonadia, Gracilibacteria, Paceibacteria, Microgenomatia, and ABY1, and Fig. 3 depicts the overlap of OTUs belonging to the phylogenetic groups detected in each sample. Chao1, Shannon, and Simpson diversity indices are shown in Table S2. The number of OTUs belonging to the five phylogenetic groups was 46 for the unfractionated sludge sample, 279 for the 0.45–0.22 μm fraction sample, and 242 for the 0.22–0.1 μm fraction sample. The majority of OTUs detected in the unfractionated sludge sample were also found in the fractionated samples. Size fractionation enabled the retrieval of present but undetected Patescibacteria in samples.
Table 1.
Numbers of OTUs in five phylogenetic groups of
Candidatus Patescibacteria
| Samples |
Saccharimonadia |
Gracilibacteria |
Paceibacteria |
Microgenomatia |
ABY1 |
Total |
| Unfractionated sludge |
16 |
18 |
9 |
1 |
2 |
46 |
| 0.45–0.22 μm fraction |
115 |
42 |
86 |
24 |
12 |
279 |
| 0.22–0.1 μm fraction |
120 |
12 |
73 |
30 |
7 |
242 |

A 16S rRNA gene-based phylogenetic tree was constructed for the five phylogenetic groups of Patescibacteria to identify the phylogenetic positions of the OTUs obtained in the present study (Fig. S1). OTUs were widely distributed among the phylogenetic groups, showing the high diversity of Patescibacteria in the activated sludge sample. The OTUs detected in the fractionated samples were phylogenetically close to those obtained from various environmental sources, such as bioreactors, soil, groundwater, and river water as well as the oral cavity. Most of the OTUs belonging to Paceibacteria, Microgenomatia, and ABY1 were phylogenetically related to those detected from size-fractionated groundwater and soil samples (pore size of 1.2–0.1 μm) (Fig. S1). Most OTUs belonging to Saccharimonadia and Gracilibacteria were phylogenetically related to those detected in bioreactor sludge samples (note that some OTUs were phylogenetically related to those detected in environmental sources, such as soil and the oral cavity). OTU781, OTU958, and OTU1753 were phylogenetically close to Saccharimonadia and were detected in activated sludge from WWTPs in Denmark (Kong et al., 2007; Albertsen et al., 2013) and France (Chouari et al., 2010). These three OTUs were only detected in the fractionated samples and were overlooked in the unfractionated sludge sample. This result indicates that various Saccharimonadia are present in activated sludge, but at a low abundance.
Recovery of metagenome-assembled patescibacterial genomes
The 0.45–0.22 μm fraction sample of activated sludge was used for further shotgun metagenomic sequencing to recover the genomes of Patescibacteria. We reconstructed 25 bins that passed the quality threshold (Table 2 and S3). Of the 25 bins, 24 belonged to Patescibacteria (one belonged to Leptospiraceae). Patescibacterial bins belonged to the taxonomic groups Paceibacteria, Saccharimonadia, ABY1, Gracilibacteria, and Microgenomatia. The expected genome size, completeness, and contamination of these bins are shown in Table 2. The bin-genome sizes recovered in the present study were similar to those in a previous study that predicted the genome size of Patescibacteria as 1.1±0.2 Mbp (Tian et al., 2020).
Table 2.
Genome size, completeness, and contamination of patescibacterial bins obtained in the present study.
|
Saccharimonadia |
Gracilibacteria |
Paceibacteria |
Microgenomatia |
ABY1 |
| Number of bins |
7 |
1 |
13 |
1 |
2 |
Average genome size
(Mbp, Min–Max) |
0.92
(0.46–1.32) |
1.09 |
0.70
(0.39–0.91) |
0.76 |
0.94
(0.93–0.94) |
Average completeness
(%, Min–Max) |
91.7
(83.7–97.7) |
88.4 |
88.9
(69.8–100) |
72.1 |
96.5
(93.0–100) |
Average contaminations
(%, Min–Max) |
2.0
(0–4.7) |
0 |
1.8
(0–7.0) |
0 |
0 |
Eighteen 16S rRNA gene sequences were retrieved from the 15 bins of Patescibacteria (Table S4). The 16S rRNA gene sequences obtained by the metagenomic analysis were phylogenetically close to the patescibacterial OTUs obtained by the amplicon analysis (Table S5); however, these OTUs were not detected or were detected with low relative abundance (i.e., 0.03–0.27%) in the unfractionated sludge sample. These results indicate that the size-fractionation approach is effective for recovering patescibacterial genomes with low abundance in activated sludge.
The construction of a genome tree revealed the phylogenetic positions of the bins obtained through the metagenomic analysis (Fig. 4). The reconstructed patescibacterial bins belonged to various families and genera, some of which were rarely found in activated sludge samples. The bins of Paceibacteria belonged to six families: UBA6899, CAIZLB01, UBA2163, UBA5272, UBA1006, and UBA9973, while those of Saccharimonadia belonged to three families: UBA7683, CAIOMD01, and AWTP1-31; however, the bins of MGA_S3 and MGA_S4 were not identified at the family level. The two bins of ABY1 belonged to different families: UBA922 and 2-12-FULL-60-25. The bins of MGA_P4 (Paceibacteria), MGA_S3, MGA_S6 (Saccharimonadia), and MGA_M1 (Microgenomatia) were phylogenetically close to the genomes recovered from the WWTPs in Germany (Schneider et al., 2021) and Denmark (Singleton et al., 2021). In contrast, the bins of Gracilibacteria and ABY1 were not phylogenetically related to any of the genomes recovered from the WWTPs.

Our approach also recovered bins belonging to patescibacterial families or genera with a small number of genomes registered in the GTDB (as of April 2022). Eight bins belonged to families with ten or fewer registered genomes (UBA 7683, CAIOMD01, and AWTP1-31 in Saccharimonadia; UBA6899 and UBA5272 in Paceibacteria), and six belonged to the genus with five or fewer registered genomes (CAJAUT01 in Microgenomatia; 2-12-FULL-41-16 in ABY1; and UBA11704, CAIXMG01, and UBA4124 in Paceibacteria).
Discussion
Although Patescibacteria is commonly present in activated sludge treating sewage, its ecophysiology and roles in sewage treatment processes remain unclear. The high microbial diversity and complexity of activated sludge ecosystems are bottlenecks for an in-depth analysis of Patescibacteria. The cells of Patescibacteria are known to be significantly small and, thus, are classified as ultramicrobacteria (cell volume, <0.1 μm3) (Luef et al., 2015; Nakai, 2020). With this characteristic, physical size fractionation has been used to enrich ultramicrobacterial and patescibacterial members in natural environmental samples. Therefore, we applied a filtration-based size-fractionation approach to an activated sludge sample and successfully enriched small coccoid-like cells (Fig. 1A).
Most of the members enriched in the size-fractionated samples were Patescibacteria, i.e., Saccharimonadia, Gracilibacteria, Paceibacteria, Microgenomatia, and ABY1, after the amplicon analysis targeting the 16S rRNA gene (Fig. 2). The number of OTUs belonging to these phylogenetic groups increased (Table 1), and a high phylogenetic diversity of Patescibacteria in activated sludge was revealed (Fig. S1). The number of patescibacterial OTUs detected in activated sludge was more than 200, which was markedly higher than those found in natural environments, such as a groundwater sample (8 OTUs) (Chik et al., 2020) and seawater sample (89 OTUs) (Suominen et al., 2021), indicating that activated sludge harbors more Patescibacteria than natural environments. Water from various environmental sources, including households, industries, and nature, is collected and sent to WWTPs, suggesting that diverse Patescibacteria have the opportunity to migrate into the system. The stable physical conditions of sewage (e.g., temperature and pH [Tian et al., 2020]) may help Patescibacteria to survive in reaction tanks. In addition, if Patescibacteria adopts a parasitic/symbiotic lifestyle as previously suggested (He et al., 2015; Moreira et al., 2021; Yakimov et al., 2022), biomass-rich activated sludge ecosystems provide more opportunities for contacting their hosts/partners than natural ecosystems, which is advantageous for the survival of Patescibacteria.
Patescibacteria have mainly been detected in anoxic or hypoxic environments (He et al., 2021), and most are considered to be anaerobes based on their metabolic capacities predicted by genome analyses (Castelle et al., 2018). In contrast, some members of Patescibacteria are known to prefer aerobic conditions, and it has been reported that the phylogenetic groups detected and their proportions depend on the oxygen concentration in the ecosystem (Herrmann et al., 2019; Chaudhari et al., 2021). Saccharimonadia are often found in aerobic and anoxic environments, as shown in the present and previous studies (Albertsen et al., 2013; Kindaichi et al., 2016; Herrmann et al., 2019; Chaudhari et al., 2021). Paceibacteria and ABY1 exhibited different preferences for oxygen at the order/family level; Candidatus Kaiserbacteraceae in Paceibacteria showed a positive correlation with oxygen concentrations, while Candidatus Nomurabacteraceae/UBA9983 in Paceibacteria and Candidatus Magasanikibacterales in ABY1 showed negative correlations (Herrmann et al., 2019). Therefore, each Patescibacteria member may find their niche in activated sludge flocs where an oxygen concentration gradient occurs.
Saccharimonadia are generally the dominant Patescibacteria in activated sludge treating sewage (Nielsen et al., 2009; Kindaichi et al., 2016) and have been clustered into at least six phylogenetic groups (G1-G6) (McLean et al., 2020). Most of the OTUs detected in the present study belonged to group G1. G1 consists of sequences derived from various environmental sources, including bioreactors and mammalian host-associated (MHA) sources with strong human relevance. Many Saccharimonadia found in sludge treating wastewater, including Ca. Saccharimonas aalborgensis (Albertsen et al., 2013), belong to this group. We found OTUs close to those detected in the activated sludge of WWTPs in other countries in the size-fractionated samples, but not in the unfractionated samples. Our in-depth analysis revealed that these saccharimonadial OTUs were not specific to each WWTP, but were commonly present in activated sludge. In addition to the characteristics of municipal wastewater and operational conditions, the ecophysiological differences in these saccharimonadial OTUs may influence which OTUs become dominant in an activated sludge ecosystem. Therefore, further studies are needed to infer physiological and ecological characteristics from saccharimonadial genomic data.
The present study revealed the high diversity of Patescibacteria in activated sludge and demonstrated that size fractionation was effective for recovering patescibacterial genomes. Further studies to elucidate the ecophysiological characteristics of previously overlooked Patescibacteria will provide novel insights into activated sludge ecosystems. The filtration-based size-fractionation approach may be applied to other sludge samples, including anaerobic sludge. In addition, because a large amount of the patescibacterial biomass may be easily recovered using this technique, the cells obtained may also be used as an inoculum source to cultivate Patescibacteria members. The acquisition of additional Patescibacteria genomes and/or enrichment cultures through the size-fractionation approach will contribute to a more detailed understanding of the ecological characteristics of Patescibacteria in wastewater treatment processes.
Citation
Kagemasa, S., Kuroda, K., Nakai, R., Li, Y.-Y., and Kubota, K. (2022) Diversity of Candidatus Patescibacteria in Activated Sludge Revealed by a Size-Fractionation Approach. Microbes Environ 37: ME22027.
https://doi.org/10.1264/jsme2.ME22027
Acknowledgements
This work was supported by Grant-in-Aids for Scientific Research (B) (KAKENHI Grant Numbers JP18H01564 and JP21H01460) from the Japan Society for the Promotion of Science (JSPS) and a Tohoku University-AIST matching fund. S. K. was supported by a Grant-in-Aid for JSPS Fellows (JP21J11654) and the MEXT WISE Program for Sustainability in Dynamic Earth (SyDE), Japan.
References
- Albertsen, M., Hugenholtz, P., Skarshewski, A., Nielsen, K.L., Tyson, G.W., and Nielsen, P.H. (2013) Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol
31: 533–538.
- Beam, P.J., Becraft, D.E., Brown, M.J., Schulz, F., Jarett, K.J., Bezuidt, O., et al. (2020) Ancestral absence of electron transport chains in Patescibacteria and DPANN. Front Microbiol
11: 1848.
- Berry, D., Ben Mahfoudh, K., Wagner, M., and Loy, A. (2011) Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl Environ Microbiol
77: 7846–7849.
- Bolger, A.M., Lohse, M., and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics
30: 2114–2120.
- Bor, B., McLean, J.S., Foster, K.R., Cen, L., To, T.T., Serrato-Guillen, A., et al. (2018) Rapid evolution of decreased host susceptibility drives a stable relationship between ultrasmall parasite TM7x and its bacterial host. Proc Natl Acad Sci U S A
115: 12277–12282.
- Brown, D.R., Whitcomb, R.F., and Bradbury, J.M. (2007) Revised minimal standards for description of new species of the class Mollicutes (division Tenericutes). Int J Syst Evol Microbiol
57: 2703–2719.
- Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K., et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods
7: 335–336.
- Castelle, C.J., Wrighton, K.C., Thomas, B.C., Hug, L.A., Brown, C.T., Wilkins, M.J., et al. (2015) Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol
25: 690–701.
- Castelle, C.J., Brown, C.T., Anantharaman, K., Probst, A.J., Huang, R.H., and Banfield, J.F. (2018) Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat Rev Microbiol
16: 629–645.
- Chaudhari, N.M., Overholt, W.A., Figueroa-Gonzalez, P.A., Taubert, M., Bornemann, T.L.V., Probst, A.J., et al. (2021) The economical lifestyle of CPR bacteria in groundwater allows little preference for environmental drivers. Environ Microbiome
16: 1–18.
- Chaumeil, P.A., Mussig, A.J., Hugenholtz, P., and Parks, D.H. (2019) GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics
36: 1925–1927.
- Chik, A.H.S., Emelko, M.B., Anderson, W.B., O’Sullivan, K.E., Savio, D., Farnleitner, A.H., et al. (2020) Evaluation of groundwater bacterial community composition to inform waterborne pathogen vulnerability assessments. Sci Total Environ
743: 140472.
- Chin, N., Pui, Y., Zainura, N.Z., Mohd, R.H., Azmi, A., Mohd, D.F.M., and Zaharah, I. (2017) Correlation between microbial community structure and performances of membrane bioreactor for treatment of palm oil mill effluent | Elsevier Enhanced Reader. Chem Eng J
305: 656–663.
- Chouari, R., Le Paslier, D., Daegelen, P., Dauga, C., Weissenbach, J., and Sghir, A. (2010) Molecular analyses of the microbial community composition of an anoxic basin of a municipal wastewater treatment plant reveal a novel lineage of Proteobacteria. Microb Ecol
60: 272–281.
- Gonzalez-Martinez, A., Rodriguez-Sanchez, A., Lotti, T., Garcia-Ruiz, M.-J., Osorio, F., Gonzalez-Lopez, J., and van Loosdrecht, M.C.M. (2016) Comparison of bacterial communities of conventional and A-stage activated sludge systems. Sci Rep
6: 18786.
- Hahn, M.W. (2003) Isolation of strains belonging to the cosmopolitan Polynucleobacter necessarius cluster from freshwater habitats located in three climatic zones. Appl Environ Microbiol
69: 5248–5254.
- He, C., Keren, R., Whittaker, M.L., Farag, I.F., Doudna, J.A., Cate, J.H.D., and Banfield, J.F. (2021) Genome-resolved metagenomics reveals site-specific diversity of episymbiotic CPR bacteria and DPANN archaea in groundwater ecosystems. Nat Microbiol
6: 354–365.
- He, X., McLean, J.S., Edlund, A., Yooseph, S., Hall, A.P., Liu, S.Y., et al. (2015) Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc Natl Acad Sci U S A
112: 244–249.
- Herrmann, M., Wegner, C.E., Taubert, M., Geesink, P., Lehmann, K., Yan, L., et al. (2019) Predominance of Cand. Patescibacteria in groundwater is caused by their preferential mobilization from soils and flourishing under oligotrophic conditions. Front Microbiol
10: 1407.
- Hosokawa, S., Kuroda, K., Narihiro, T., Aoi, Y., Ozaki, N., Ohashi, A., and Kindaichi, T. (2021) Cometabolism of the superphylum Patescibacteria with anammox bacteria in a long-term freshwater anammox column reactor. Water (Basel, Switz)
13: 208.
- Hu, M., Wang, X., Wen, X., and Xia, Y. (2012) Microbial community structures in different wastewater treatment plants as revealed by 454-pyrosequencing analysis. Bioresour Technol
117: 72–79.
- Hugenholtz, P., Tyson, G.W., Webb, R.I., Wagner, A.M., and Blackall, L.L. (2001) Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives. Appl Environ Microbiol
67: 411–419.
- Ju, F., and Zhang, T. (2015) Bacterial assembly and temporal dynamics in activated sludge of a full-scale municipal wastewater treatment plant. ISME J
9: 683–695.
- Kang, D.D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., and Wang, Z. (2019) MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ
7: e7359.
- Kindaichi, T., Yamaoka, S., Uehara, R., Ozaki, N., Ohashi, A., Albertsen, M., et al. (2016) Phylogenetic diversity and ecophysiology of Candidate phylum Saccharibacteria in activated sludge. FEMS Microbiol Ecol
92: fiw078.
- Kong, Y., Xia, Y., Nielsen, J.L., and Nielsen, P.H. (2007) Structure and function of the microbial community in a full-scale enhanced biological phosphorus removal plant. Microbiology
153: 4061–4073.
- Li, D., Liu, C.M., Luo, R., Sadakane, K., and Lam, T.W. (2015) MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics
31: 1674–1676.
- Li, D., Luo, R., Liu, C.M., Leung, C.M., Ting, H.F., Sadakane, K., et al. (2016) MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods (Amsterdam, Neth)
102: 3–11.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics
25: 2078–2079.
- Li, X., Deng, Y., Li, X., Ma, X., Wang, J., and Li, J. (2020) Integration of marine macroalgae (Chaetomorpha maxima) with a moving bed bioreactor for nutrient removal from maricultural wastewater. Archaea
2020: 1–13.
- Lin, H.H., and Liao, Y.C. (2016) Accurate binning of metagenomic contigs via automated clustering sequences using information of genomic signatures and marker genes. Sci Rep
6: 24175.
- Ludington, W.B., Seher, T.D., Applegate, O., Li, X., Kliegman, J.I., Langelier, C., et al. (2017) Assessing biosynthetic potential of agricultural groundwater through metagenomic sequencing: A diverse anammox community dominates nitrate-rich groundwater. PLoS One
12: e0174930.
- Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, et al. (2004) ARB: a software environment for sequence data. Nucleic Acids Res
32: 1363–1371.
- Luef, B., Frischkorn, K.R., Wrighton, K.C., Holman, H.Y., Birarda, G., Thomas, B.C., et al. (2015) Diverse uncultivated ultra-small bacterial cells in groundwater. Nat Commun
6: 6372.
- Martínez-Campos, S., González-Pleiter, M., Fernández-Piñas, F., Rosal, R., and Leganés, F. (2021) Early and differential bacterial colonization on microplastics deployed into the effluents of wastewater treatment plants. Sci Total Environ
757: 143832.
- Matsubayashi, M., Shimada, Y., Li, Y.Y., Harada, H., and Kubota, K. (2017) Phylogenetic diversity and in situ detection of eukaryotes in anaerobic sludge digesters. PLoS One
12: e0172888.
- McLean, J.S., Bor, B., Kerns, K.A., Liu, Q., To, T.T., Solden, L., et al. (2020) Acquisition and adaptation of ultra-small parasitic reduced genome bacteria to mammalian hosts. Cell Rep
32: 107939.
- Minh, B.Q., Schmidt, H.A., Chernomor, O., Schrempf, D., Woodhams, M.D., von Haeseler, A., and Lanfear, R. (2020) IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol
37: 1530–1534.
- Moreira, D., Zivanovic, Y., López-Archilla, A.I., Iniesto, M., and López-García, P. (2021) Reductive evolution and unique predatory mode in the CPR bacterium Vampirococcus lugosii. Nat Commun
12: 2454.
- Nakai, R. (2020) Size matters: Ultra-small and filterable microorganisms in the environment. Microbes Environ
35: ME20025.
- Ni, J., Hatori, S., Wang, Y., Li, Y.Y., and Kubota, K. (2020) Uncovering viable microbiome in anaerobic sludge digesters by propidium monoazide (PMA)-PCR. Microb Ecol
79: 925–932.
- Nielsen, P.H., Kragelund, C., Seviour, R.J., and Nielsen, J.L. (2009) Identity and ecophysiology of filamentous bacteria in activated sludge. FEMS Microbiol Rev
33: 969–998.
- Nierychlo, M., Andersen, K., Xu, Y., Green, N., Jiang, C., Albertsen, M., et al. (2020) MiDAS 3: An ecosystem-specific reference database, taxonomy and knowledge platform for activated sludge and anaerobic digesters reveals species-level microbiome composition of activated sludge. Water Res
182: 115955.
- Nissen, J.N., Johansen, J., Allesøe, R.L., Sønderby, C.K., Armenteros, J.J., Grønbech, C.H., et al. (2021) Improved metagenome binning and assembly using deep variational autoencoders. Nat Biotechnol
39: 555–560.
- Olm, M.R., Brown, C.T., Brooks, B., and Banfield, J.F. (2017) dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J
11: 2864–2868.
- Parks, D.H., Imelfort, M., Skennerton, C.T., Hugenholtz, P., and Tyson, G.W. (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res
25: 996–1004.
- Pernthaler, A., Pernthaler, J., and Amann, R. (2002) Fluorescence in situ hybridization and catalyzed reporter deposition for the identification of marine bacteria. Appl Environ Microbiol
68: 3094–3101.
- Probst, A.J., Castelle, C.J., Singh, A., Brown, C.T., Anantharaman, K., Sharon, I., et al. (2017) Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO2 concentrations. Environ Microbiol
19: 459–474.
- Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res
41: D590–D596.
- Remmas, N., Melidis, P., Zerva, I., Kristoffersen, J.B., Nikolaki, S., Tsiamis, G., and Ntougias, S. (2017) Dominance of candidate Saccharibacteria in a membrane bioreactor treating medium age landfill leachate: Effects of organic load on microbial communities, hydrolytic potential and extracellular polymeric substances. Bioresour Technol
238: 48–56.
- Rendulic, S., Jagtap, P., Rosinus, A., Eppinger, M., Baar, C., Lanz, C., et al. (2004) A predator unmasked: life cycle of Bdellovibrio bacteriovorus from a genomic perspective. Science
303: 689–692.
- Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016) VSEARCH: a versatile open source tool for metagenomics. PeerJ
4: e2584.
- Schneider, D., Zühlke, D., Poehlein, A., Riedel, K., and Daniel, R. (2021) Metagenome-assembled genome sequences from different wastewater treatment stages in Germany. Microbiol Resour Announce
10: 863–864.
- Seemann, T. (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics
30: 2068–2069.
- Sekiguchi, Y., Ohashi, A., Parks, D.H., Yamauchi, T., Tyson, G.W., and Hugenholtz, P. (2015) First genomic insights into members of a candidate bacterial phylum responsible for wastewater bulking. PeerJ
3: e740.
- Sieber, C.M.K., Probst, A.J., Sharrar, A., Thomas, B.C., Hess, M., Tringe, S.G., and Banfield, J.F. (2018) Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol
3: 836–843.
- Singleton, C.M., Petriglieri, F., Kristensen, J.M., Kirkegaard, R.H., Michaelsen, T.Y., Andersen, M.H., et al. (2021) Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing. Nat Commun
12: 2009.
- Suominen, S., Doorenspleet, K., Sinninghe Damsté, J.S., and Villanueva, L. (2021) Microbial community development on model particles in the deep sulfidic waters of the Black Sea. Environ Microbiol
23: 2729–2746.
- Tian, R., Ning, D., He, Z., Zhang, P., Spencer, S.J., Gao, S., et al. (2020) Small and mighty: adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome
8: 126085.
- Tully, B.J., Graham, E.D., and Heidelberg, J.F. (2018) The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci Data
5: 170203.
- Vigneron, A., Lovejoy, C., Cruaud, P., Kalenitchenko, D., Culley, A., and Vincent, W.F. (2019) Contrasting winter versus summer microbial communities and metabolic functions in a permafrost thaw lake. Front Microbiol
10: 1656.
- Wang, P., Yu, Z., Zhao, J., and Zhang, H. (2018) Do microbial communities in an anaerobic bioreactor change with continuous feeding sludge into a full-scale anaerobic digestion system?
Bioresour Technol
249: 89–98.
- Wrighton, K.C., Thomas, B.C., Sharon, I., Miller, C.S., Castelle, C.J., VerBerkmoes, N.C., et al. (2012) Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science
337: 1661–1665.
- Wu, Y.W., Tang, Y.H., Tringe, S.G., Simmons, B.A., and Singer, S.W. (2014) MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome
2: 1183–1195.
- Xu, S., Yao, J., Ainiwaer, M., Hong, Y., and Zhang, Y. (2018) Analysis of bacterial community structure of activated sludge from wastewater treatment plants in winter. BioMed Res Int
2018: 8278970.
- Yakimov, M.M., Merkel, A.Y., Gaisin, V.A., Pilhofer, M., Messina, E., Hallsworth, J.E., et al. (2022) Cultivation of a vampire: ‘Candidatus Absconditicoccus praedator’. Environ Microbiol
24: 30–49.
- Zhang, T., Shao, M.F., and Ye, L. (2012) 454 pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J
6: 1137–1147.