Abstract
Patescibacteria are widely distributed in various environments and often detected in activated sludge. However, limited information is currently available on their phylogeny, morphology, and ecophysiological role in activated sludge or interactions with other microorganisms. In the present study, we identified microorganisms that interacted with Patescibacteria in activated sludge via a correlation analysis using the 16S rRNA gene, and predicted the metabolic potential of Patescibacteria using a metagenomic analysis. The metagenome-assembled genomes of Patescibacteria consisted of three Saccharimonadia, three Parcubacteria, and one Gracilibacteria, and showed a strong positive correlation of relative abundance with Chitinophagales. Metabolic predictions from ten recovered patescibacterial and five Chitinophagales metagenome-assembled genomes supported mutualistic interactions between a member of Saccharimonadia and Chitinophagales via N-acetylglucosamine, between a member of Parcubacteria and Chitinophagales via nitrogen compounds related to denitrification, and between Gracilibacteria and Chitinophagales via phospholipids in activated sludge. The present results indicate that various interactions between Patescibacteria and Chitinophagales are important for the survival of Patescibacteria in activated sludge ecosystems.
The phylogeny and physiology of microorganisms in activated sludge for wastewater treatment remain unclear due to their complexity and variations even though the activated sludge process has been used in wastewater treatment globally for more than 100 years. The structure of a microbial community depends on the climate, location, environment, and process configuration of the wastewater treatment plant (Zhang et al., 2012). Recent studies reported that communities appeared to be stable at the genus and substrate specificity levels (Nielsen et al., 2010; Kindaichi et al., 2013). Most microorganisms, including those in nature and in engineered systems, cannot be cultured in laboratories and are called “microbial dark matter” (Rinke et al., 2013). In this context, molecular biological methods, such as a 16S rRNA gene analysis and metagenomic analysis, have been widely used to predict the metabolic functions of uncultured bacteria. Large metagenomic data obtained from samples in various environments revealed the existence of a large group of bacteria, called Patescibacteria or candidate phyla radiation (CPR) (Rinke et al., 2013; Brown et al., 2015). The Patescibacteria or CPR (hereafter called Patescibacteria) group includes 35 phyla, accounts for 15–50% of all bacterial phyla, and has been reported to exist in various environments (Brown et al., 2015; Hug et al., 2016; Takebe et al., 2020). To date, only a few of its members (i.e., phyla Saccharimonadia and Gracilibacteria) have been cultured (Soro et al., 2014; He et al., 2015; Ibrahim et al., 2021; Yakimov et al., 2021). These bacteria are parasitic on other bacteria for sustenance; however, since most of them cannot be cultured, the mechanisms underlying their existence are unclear. Patescibacteria are commonly characterized by a small genome size (approximately 1.0 Mbp) (Lemos et al., 2020; Nakai, 2020), limited metabolic potential, and fermentation-based metabolism (Wrighton et al., 2012, 2014; Albertsen et al., 2013; Lemos et al., 2020). However, the physiology and phylogeny of Patescibacteria have not yet been elucidated in detail, except for some cultures in the phyla Saccharimonadia and Gracilibacteria (Soro et al., 2014; He et al., 2015; Moreira et al., 2021; Yakimov et al., 2021).
Activated sludge is an environment in which Patescibacteria are frequently detected. Among them, Saccharimonadia, Parcubacteria, and Gracilibacteria are the major phyla (Albertsen et al., 2013; Kindaichi et al., 2016; Singleton et al., 2021). A moderate constituent of activated sludge is Saccharimonadia, a well-described Patescibacteria (Mielczarek et al., 2012; Albertsen et al., 2013; Kindaichi et al., 2016). Based on the 16S rRNA gene classification, Saccharimonadia are primarily classified into three subdivisions, with members having a filamentous morphology belonging to subdivision 1, and members with a coccus or rod morphology belonging to subdivisions 2 and 3 (Hugenholtz et al., 2001). A complete genome belonging to subdivision 3 was reconstructed from activated sludge samples and the data obtained showed that Saccharimonadia are obligate fermentative metabolic bacteria that use heterolactic fermentation pathways (Albertsen et al., 2013). In addition, filamentous Saccharimonadia were detected in activated sludge from wastewater treatment plants, and the characteristics of substrate utilization elucidated by microautoradiography combined with fluorescence in situ hybridization (FISH) revealed more diverse carbon metabolism, including the utilization of oleic acid and amino acids, which was not predicted from the available genome (Kindaichi et al., 2016). Parcubacteria also belong to Patescibacteria and are found in activated sludge (Zhang et al., 2012). Parcubacteria have a diverse distribution within the phylum, with most members being frequently found in anaerobic environments, such as groundwater. Parcubacteria are considered to be involved in hydrogen production, sulfur reduction, and nitrite reduction (Wrighton et al., 2012; 2014; Rinke et al., 2013; Danczak et al., 2017). Additionally, a syntrophic relationship with other bacteria has been suggested as a putative benzene degrader in anaerobic environments (Phan et al., 2021). However, some members of Parcubacteria harbor genes that are capable of using O2 as a terminal electron acceptor (Nelson and Stegen, 2015). Gracilibacteria include three lineages and belong to Patescibacteria. Hanke et al. predicted that the terminal codon UGA encodes glycine in Gracilibacteria (Hanke et al., 2014). These bacteria have poor metabolic potential (Sieber et al., 2019), and some strains were reported to be parasitic on their hosts (Moreira et al., 2021; Yakimov et al., 2021).
Although many high-quality genomes related to Patescibacteria have been obtained from various environments, their detailed phylogeny, morphology, and ecophysiological role in activated sludge remain largely unknown. To clarify the phylogenetic and physiological diversities of Patescibacteria in activated sludge, obtaining high-quality genomes of Patescibacteria is necessary for further investigations in terms of visualization, in situ substrate utilization, and isolation. The purpose of the present study was to predict the metabolic potential of Patescibacteria in activated sludge and estimate their physiological role in activated sludge. A metagenomic approach using three activated sludge samples from a municipal wastewater treatment plant recovered 10 metagenome-assembled genomes (MAGs) related to Saccharimonadia, Parcubacteria, and Gracilibacteria within the superphylum Patescibacteria.
Materials and Methods
Sample collection
Four activated sludge samples were collected from aeration tanks in a wastewater treatment plant in Higashihiroshima city, which had previously been sampled (Kindaichi et al., 2016; Table S1) in February 2019 (designated as AS201902), April 2020 (designated as AS202004), October 2020 (designated as AS202010R), and November 2020 (designated as AS202011). The collected sludge samples were immediately incubated to change the relative abundance of Patescibacteria. The AS202004 sample was anaerobically incubated for 3 d and was then designated as AA202004. The AS202010R sample was aerobically incubated for 3 d with washing and designated as AS202010A and without washing as AS202010B. In detail, 100 mL of the AS202004 sample was transferred into a 120-mL sterilized vial, which was sealed with a butyl rubber stopper. The gas phase was replaced with nitrogen gas, and the vial was then incubated anaerobically at 20°C for 3 d. One hundred milliliters of activated sludge from sample AS202010R was washed with Elix water (Merck) and then incubated at 20°C for 3 d. In the present study, the samples AS201902, AS202004, and AA202004 were used in a metagenomic analysis, while all seven samples were subjected to an amplicon analysis. Fresh and incubated sludge samples were stored at –18°C for further analyses.
Amplicon analysis of the 16S rRNA gene
DNA was extracted from activated sludge samples (0.5 g wet weight) (AS201902, AS202004, AA202004, AS202010A, AS202010B, AS202010, and AS202011) using a FastDNA SPIN kit for soil (MP Biomedicals). PCR amplification was performed using a primer set for the V3–V4 region of the 16S rRNA genes (341F and 805R). The primer sequences, detailed PCR conditions, and purification procedures used are as previously described (Dinh et al., 2021). Purified DNA was sequenced using a MiSeq platform with paired-end sequencing (2×300 bp) and a MiSeq Reagent kit (v.3; Illumina). The obtained sequences were trimmed, merged, clustered, and analyzed using QIIME 2 core 2021.11, as previously described (Bolyen et al., 2019; Awata et al., 2021; Kambara et al., 2022). The SILVA 138 database (Quast et al., 2013) was used for the assignment. To elucidate the relationship between Patescibacteria and other co-existing bacteria, operational taxonomic units (OTUs) that showed a relative abundance of >0.1% were extracted, and Spearman’s rank-order correlation coefficient was calculated for each OTU using Past 4.10 (Hammer et al., 2001). OTUs that met the 5% significance level and correlated with Patescibacteria were investigated.
Metagenomic analysis
DNA was extracted from activated sludge samples (0.5 g wet weight) (AS201902, AS202004, and AA202004) using a FastDNA SPIN kit for soil (MP Biomedicals). Extracted DNA was purified using Agencourt AMPure XP magnetic beads (Beckman Coulter Life Sciences). Illumina sequencing libraries were prepared for the three samples using a TruSeq DNA PCR Free (350) kit (Illumina) and paired-end sequenced (2×151 bp) using shotgun sequencing on a HiSeq X system (Illumina). PacBio sequencing libraries were prepared for three samples using a 20 kb SMRTbell Express Template Prep kit (Pacific Biosciences of California) and sequenced on a PacBio Sequel II System (Pacific Biosciences of California). Circular consensus sequence (CCS) reads were generated from Sequel data with a Phred quality score above 20 (Q20, 99%).
A metagenomic analysis was conducted as previously described (Hosokawa et al., 2021). Raw paired-end reads from HiSeq X were trimmed using Trimmomatic v.0.39 (Bolger et al., 2014). The trimmed reads from HiSeq X and CCS reads from PacBio Sequel II were co-assembled using SPAdes v.3.13.1 (Bankevich et al., 2012). BBtools v38.84 was used to obtain mapping information. Contigs from the assembly were binned using MetaBAT2.0 (Kang et al., 2019). The relative abundance of the bins (multi-contigs classified into a taxonomic microorganism) were calculated based on information from the mapping file (i.e., coverage) generated in MetaBAT2.0. The completeness and contamination of the bins were assessed using CheckM v1.1.2 (Parks et al., 2015). The 43 marker genes proposed by Brown et al. (2015) likely provide improved estimates of CPR genome quality. Contamination in the obtained bins was manually removed. The bins with contamination removed were annotated using Prokka v1.13 (Seemann, 2014) and DRAM v1.2.2 (Shaffer et al., 2020). Predicted amino acid sequences were annotated using the KEGG BlastKEGG Orthology And Links Annotation (BlastKOALA) (Kanehisa et al., 2016) and KEGG Automatic Annotation Server (KAAS) (Moriya et al., 2007). BlastKOALA was used to visualize this pathway. A heatmap was created using KEGG-Decoder (Graham et al., 2018) to visualize the percentage of gene possession related to each gene set. A phylogenetic tree of Patescibacteria, based on 400 marker protein sequences, was constructed using Phylophlan 3.0 (Asnicar et al., 2020). The reference genome was selected from the genome registered in GenBank, and the complete genome was derived from activated sludge (Singleton et al., 2021). Polyhydroxybutyrate (PHB) depolymerase-related sequences were aligned using mafft-linsi v7.480 (default parameters) (Katoh and Standley, 2013). Reference protein sequences were obtained from the top 500 hits for the identified PHB depolymerase-related protein (FNKGEGDK_00198) and known patescibacterial PHB depolymerase (OWK27304.1) using the NCBI-nr database. Protein sequences were clustered based on ≥70% similarity using CD-HIT version 4.8.1. (Fu et al., 2012). A phylogenetic tree of PHB depolymerase-related proteins was constructed using iqtree2 version 2.1.2, with an automatically optimized substitution model of WAG+R10 (Minh et al., 2020).
Nucleotide sequence accession number
The sequence data of the partial 16S rRNA gene sequence were deposited in the GenBank/EMBL/DDBJ databases under the accession number DRA013509. Metagenomic sequence data were deposited in the DDBJ database under the DDBJ/EMBL/GenBank accession number DRA013531.
Results and Discussion
Amplicon analysis of 16S rRNA genes
Amplicon sequencing of the 16S rRNA genes was performed to investigate the relative abundance of Patescibacteria in the seven activated sludge samples. On average, 34,573 reads and 549 OTUs were obtained from the seven samples (Table S3). In all activated sludge samples, except AS202004, Patescibacteria were predominant after Proteobacteria and Bacteroidota, with an average abundance of 12.1% (Fig. 1A). The most dominant group within Patescibacteria was Saccharimonadia in all samples, with the highest abundance of 13.7% in AS202010R. Parcubacteria and Gracilibacteria were the second and third most abundant groups, respectively (Fig. 1B). In addition to the above-mentioned groups, Microgenomatia (former candidate division OP11), ABY1, Dojkabacteria (former candidate division WS6), and Berkelbacteria were detected; however, their relative abundance was less than 0.2%. The ranges of the relative abundance of Saccharimonadia, Parcubacteria, and Gracilibacteria in untreated activated sludge samples (AS201902, AS202004, AS202010R, and AS202011) were 4.5–13.7%, 2.4–5.0%, and 0.4–1.8%, respectively. The relative abundance of Patescibacteria in all three treated samples (i.e., aerobic and anaerobic incubations) decreased (Fig. S1). The relative abundance of Saccharimonadia, Parcubacteria, and Gracilibacteria in AS202004 were 9.5, 3.5, and 1.1%, respectively, whereas those in AA202004 were 5.5, 2.8, and 0.6%, respectively. The relative abundance of Saccharimonadia, Parcubacteria, and Gracilibacteria in AS202010R, AS202010A, and AS202010B were 9.5, 3.5, and 1.1%, 5.5, 2.8, and 0.6%, and 5.5, 2.8, and 0.6%, respectively. This decrease may be associated with the oxygen level or abundance of coexisting bacteria. The relative abundance of Patescibacteria in groundwater samples ranged between 2.1 and 20.7%; however, it was not possible to compare these samples directly because they were enriched using a filter-based sampling method (Danczak et al., 2017).
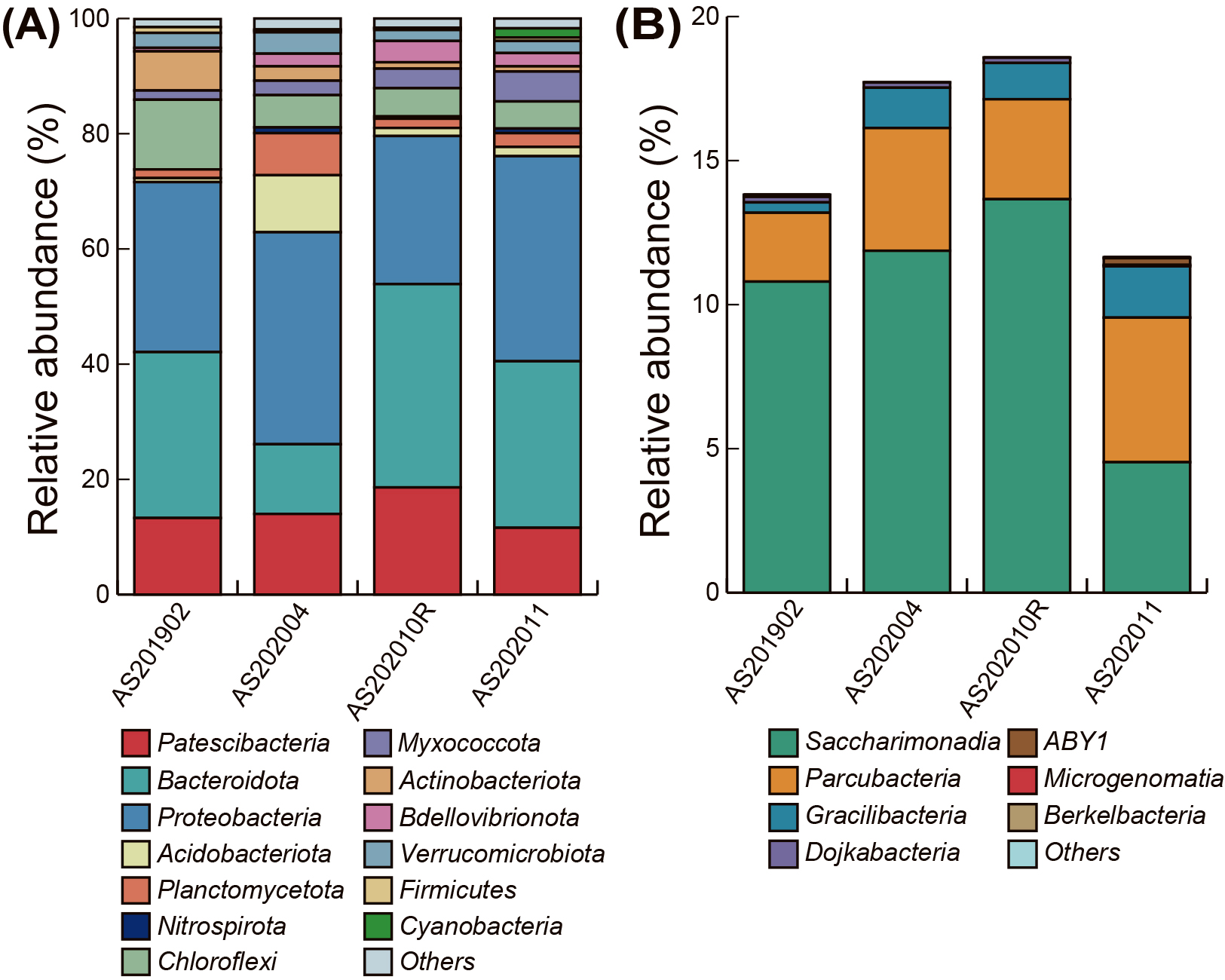
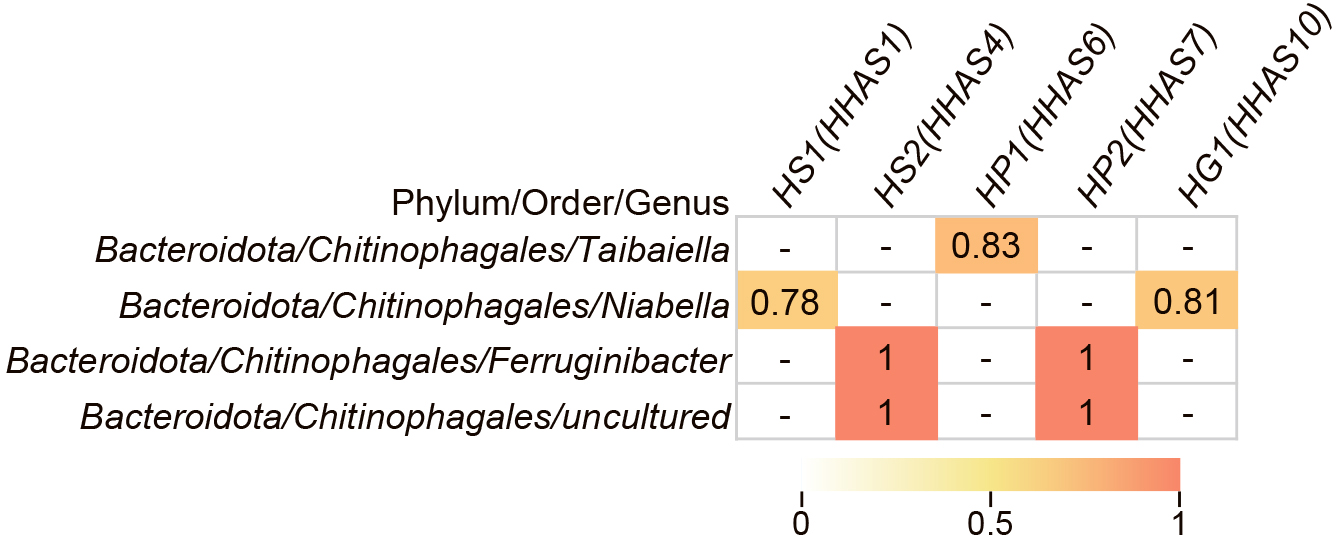
Pearson’s correlation coefficients were calculated between the patescibacterial OTUs obtained from amplicon sequencing and other bacterial OTUs with more that 0.1% relative abundance. The OTUs of Proteobacteria, Chloroflexi, and Planctomycetota showed a positive correlation with the OTUs of Patescibacteria; however, in some cases, correlations were negative (data not shown). In contrast, the correlation between Patescibacteria and Chitinophagales belonging to the phylum Bacteroidota was positive (Fig. S2). Among the OTUs shown in Fig. S2, we extracted OTUs with a sequence that matched the reconstructed bin with 100% sequence identity (Fig. 2). Most Chitinophagales OTUs correlated with several patescibacterial OTUs (HS1, HS2, and HP2). In addition, two Chitinophagales OTUs correlated with the three lineages of Patescibacteria. A similar positive correlation between Saccharimonadia and Chitinophagaceae was found in acid mine drainage samples (Lemos et al., 2019). Metabolic interactions between Patescibacteria and Chitinophagales are discussed in the following section.
In total, 0.77 billion reads and 0.06 million reads were obtained from HiSeq X and PacBio CCS sequencing of the three activated sludge samples, respectively (Table S2). The hybrid assembly using HiSeq X and PacBio CCS reads generated 12,097 contigs with an N50 value of 148,787 bp. A total of 8,211 contigs >1,500 bp were extracted and classified into 320 bins. Ten patescibacterial bins were reconstructed, which consisted of Saccharimonadia (five bins, HHAS1–HHAS5), Parcubacteria (four bins, HHAS6–HHAS9), and Gracilibacteria (one bin, HHAS10) (Table 1). The completeness of Sacchrimonadia and Parcubacteria ranged between 88.4 and 97.7 and between 62.8 and 90.7%, respectively, while that of Gracilibacteria was 97.7%.
Table 1.
Characteristics of patescibacterial bins obtained in the present study
| Bin ID |
Taxonomy |
Bin size (Mbp) |
Completeness (%) |
Contamination (%) |
Number of contigs |
Number of CDSs |
Relative abundance
(%)† |
| AS201902 |
AS202004 |
AA202004 |
| HHAS1 |
Saccharimonadia |
0.91 |
88.37* |
0* |
3 |
946 |
1.79 |
0.04 |
0.03 |
| HHAS2 |
Saccharimonadia |
0.83 |
97.67* |
0* |
3 |
847 |
0.20 |
0 |
0 |
| HHAS3 |
Saccharimonadia |
1.00 |
93.02* |
0* |
2 |
1027 |
1.47 |
0 |
0 |
| HHAS4 |
Saccharimonadia |
0.73 |
90.70* |
0* |
3 |
759 |
2.26 |
0 |
0 |
| HHAS5 |
Saccharimonadia |
0.71 |
88.37* |
0* |
6 |
746 |
0.29 |
0 |
0 |
| HHAS6 |
Parcubacteria |
0.53 |
90.70* |
0* |
8 |
536 |
0.15 |
0 |
0 |
| HHAS7 |
Parcubacteria |
0.60 |
62.79* |
0* |
2 |
627 |
0.48 |
0 |
0 |
| HHAS8 |
Parcubacteria |
0.60 |
90.70* |
0* |
5 |
619 |
0.02 |
0.26 |
0.19 |
| HHAS9 |
Parcubacteria |
0.96 |
79.07* |
0* |
6 |
942 |
0.29 |
0.27 |
0.44 |
| HHAS10 |
Gracilibacteria |
1.30 |
97.67* |
0* |
3 |
1175 |
0.02 |
0.22 |
0.10 |
| HHAS11 |
Chitinophagales |
2.37 |
75.24 |
0 |
17 |
2051 |
0 |
0.17 |
0.23 |
| HHAS12 |
Chitinophagales |
2.79 |
75.2 |
3.96 |
21 |
2435 |
0.97 |
0 |
0.02 |
| HHAS13 |
Chitinophagales |
3.13 |
94.77 |
3.45 |
5 |
2669 |
1.85 |
0 |
0 |
| HHAS14 |
Chitinophagales |
2.99 |
91.21 |
2.72 |
14 |
2354 |
0.78 |
0 |
0 |
* Calculated using the CPR marker set.
† Calculated based on the mapping file generated in MetaBAT2.0
A phylogenetic tree of the ten bins based on the protein sequence is shown in Fig. 3. Bins belonging to Saccharimonadia were classified into three groups. The group including HHAS3 and HHAS4 was related to the well-described saccharimonadial species Candidatus Saccharimonas aalborgensis (CP005957), reconstructed from a Danish activated sludge sample (Albertsen et al., 2013). Since this species shows a small coccus morphology (Albertsen et al., 2013), HHAS3 and HHAS4 were also considered to be small cocci. The HHAS1 and HHAS5 groups were related to the genomes of activated sludge samples. According to sequence similarities based on 16S rRNA genes, the morphology of this group was primarily filamentous (Kindaichi et al., 2016). The morphology of filamentous Saccharimonadia needs to be confirmed using FISH in the future. The HHAS2 bin formed a different clade from other saccharimonadial genomes with a genome from activated sludge (Singleton et al., 2021). However, the details of this group remain largely unknown. The parcubacterial bins were classified into three groups. The HHAS7 bin was classified as Nomurabacteria and was related to genomes from groundwater samples (Brown et al., 2015). Groups HHAS6 and HHAS9 belonged to Moranbacteria. In addition, clades HHAS6 and HHAS9 consisted of genomes from activated sludge samples (Singleton et al., 2021). The details of Moranbacteria in activated sludge samples are also unclear because the majority of information on Moranbacteria was obtained from groundwater samples (Anantharaman et al., 2016). The HHAS8 bin did not belong to any parcubacterial subgroup. HHAS10 was classified as belonging to Gracilibacteria. Although some of the gracilibacterial genomes were also reconstructed from activated sludge samples (Singleton et al., 2021), the HHAS10 bin formed a clade that included genomes from human oral samples (Dudek et al., 2017).

The predicted metabolic potential of Gracilibacteria, Parcubacteria, and Saccharimonadia, and the putative metabolic interactions between Patescibacteria and Chitinophagales based on the metagenomic analysis in this study are shown in Fig. 4. Patescibacterial bins revealed that Patescibacteria did not possess de novo nucleotide synthesis, amino acid synthesis, phospholipid synthesis, or a full TCA cycle. In addition, Patescibacteria possessed ABC transporters with unknown functions, the peptidoglycan biosynthesis pathway, and type IV pili (Fig. S3). The lack of de novo amino acid synthesis suggests that peptidases acquire amino acids. The presence of peptidases was also confirmed (Table S4). Several patescibacterial bins converted glycine to serine and harbored serine peptidases. These common features are consistent with the genomes of activated sludge samples as well as other natural samples (Wrighton et al., 2012, 2014; Albertsen et al., 2013; Danczak et al., 2017; Starr et al., 2018; Lemos et al., 2019; 2020; Sieber et al., 2019; Chaudhari et al., 2021; Moreira et al., 2021; Yakimov et al., 2021). The incomplete nucleotide synthesis pathway and the presence of the comE gene and type IV pili support the acquisition of DNA from outside cells (Chen and Gotschlich, 2001; Starr et al., 2018).
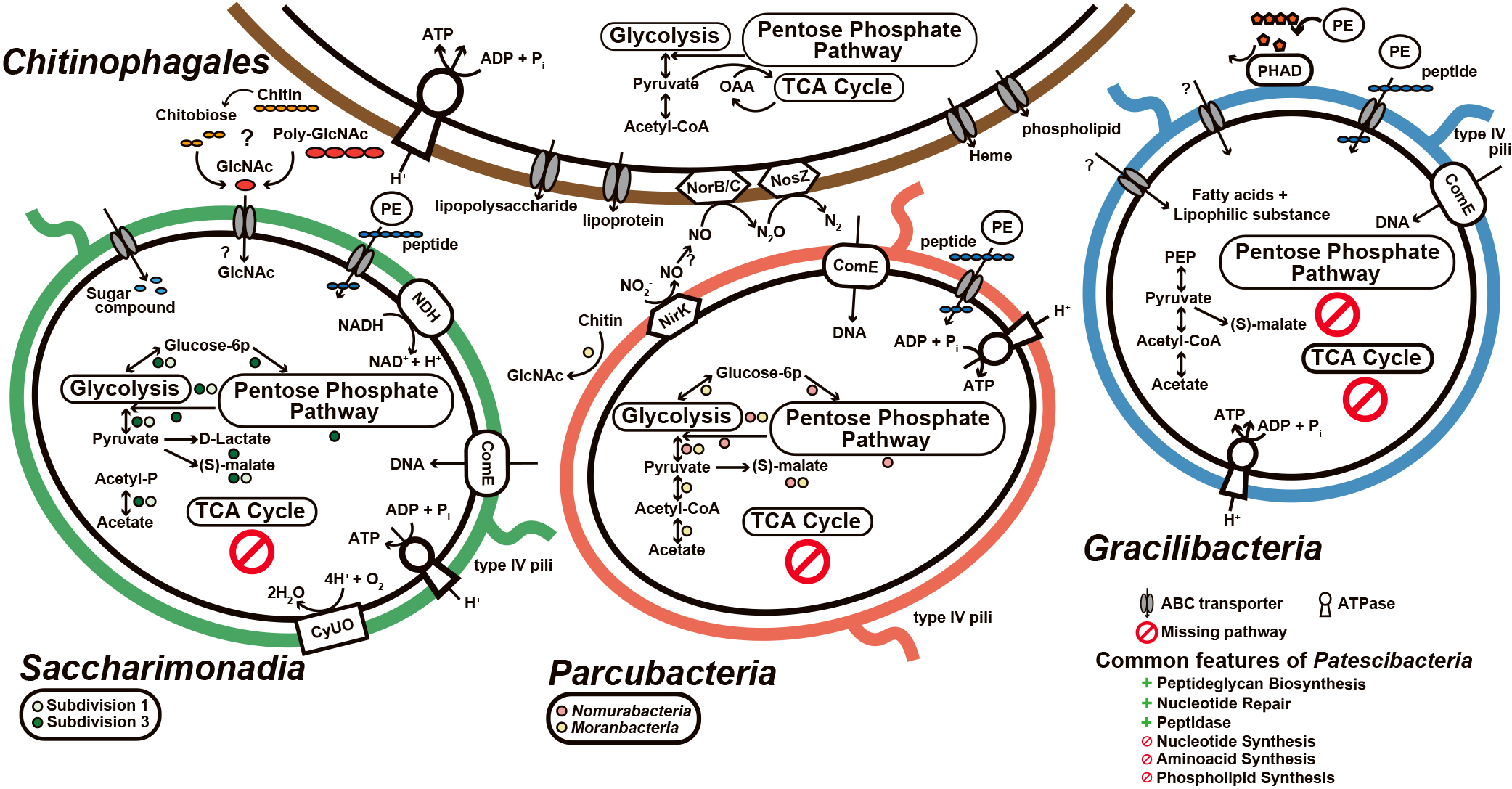
Saccharimonadial bins possessed glycolysis and the pentose phosphate pathway, with possession patterns depending on the subgroup (Fig. 4). The members of subdivision 1 (HHAS1 and HHAS5), which are putative filamentous Saccharimonadia, partially possessed glycolysis, whereas members of subdivision 3 (HHAS3 and HHAS4) possessed both glycolysis and the pentose phosphate pathway. The possession of genes to convert pyruvate to lactate, acetate, and malate and the lack of a TCA cycle supports fermentative metabolism. These results are consistent with previous findings (Wrighton et al., 2012, 2014; Albertsen et al., 2013; Danczak et al., 2017; Starr et al., 2018; Lemos et al., 2019; 2020; Sieber et al., 2019; Moreira et al., 2021; Yakimov et al., 2021). The fermentative pathway from pyruvate to lactate or malate may facilitate the production of NAD+ (Starr et al., 2018; Lemos et al., 2019). The pentose phosphate pathway in subdivision 3 may be involved in the conversion of glucose-6P to glyceradehyde-3P and in energy conversion (NADPH to NADP production) (Albertsen et al., 2013). However, genes involved in the synthesis of nucleic acids, such as ribose-phosphate pyrophosphokinase, were absent. Therefore, they are not expected to contribute to anabolism (the production of deoxyribonucleotides) (Castelle et al., 2018). All reconstructed saccharimonadial bins in the present study possessed the NADH dehydrogenase-like protein and complete cytochrome o ubiquinol oxidase, which is related to the oxygen scavenging system, despite the absence of the TCA cycle (Kantor et al., 2013; Starr et al., 2018; Lemos et al., 2019). In addition, Lemos et al. (2019) suggested that Saccharimonadia follow non-obligatory fermentative metabolism with occasional aerobic respiration. As previously reported by Lemos et al. (2019), Saccharimonadia have membrane-bound NADH dehydrogenase to supply NAD+ and pass the electron to ubiquinone, which transfers it to cytochrome O ubiquinol oxidase. Cytochrome then reduces O2 to H2O as the final receptor, delivering protons through the plasma membrane to generate the proton electromotive force used for ATP synthesis by ATP synthase. Filamentous Saccharimonadia in activated sludge took up N-acetylglucosamine under aerobic conditions, as demonstrated by microautoradiography combined with FISH (Kindaichi et al., 2016). Chitinophagales bins (HHAS12, HHAS13, and HHAS14) possessed chitinase (MHHEHLFG_00073, MHHEHLFG_01825, AECFEMFL_01184, and KHEBLPDM_00108) and all Chitinophagales bins harbored beta-acetylhexosamidase (MHHEHLFG_00810, CLEEFKKN_01010, AECFEMFL_02009, and KHEBLPDM_01401). Chitinophagales have the potential to convert chitin to N-acetylglucosamine via chitobiose (Fig. 4). All Chitinophagales bins encoded poly-beta-1,6 N-acetyl-d-glucosamine synthase (PgaC). This enzyme catalyzes the polymerization of uridine diphosphate-N-acetylglucosamine to produce poly-N-acetylglucosamine (PGA). There were other bins in active sludge belonging to Ignavibacteria, Acidobacteria, Actinobacteria, Bacteroidota,
Chloroflexi, Nitrospira, Proteobacteria, and Verrucomicrobia. These bacteria possessed PgaC and were present in approximately 38–41% of samples from AS201902, AS202004, and AA202004. Filamentous Saccharimonadia in activated sludge took up N-acetylglucosamine, which strongly supports the metabolic interaction between Chitinophagales and Saccharimonadia via N-acetylglucosamine in activated sludge. In addition, Saccharimonadia have been suggested to use some of the PGA produced by bacteria (Hosokawa et al., 2021). The mechanisms by which N-acetylglucosamine is assimilated or catabolized by Saccharimonadia currently remain unclear. Investigations on the metabolism of incorporated N-acetylglucosamine are highly challenging, but are warranted.
Parcubacterial bins possessed glycolysis and/or the pentose phosphate pathway. Parcubacterial bins also encoded genes involved in the conversion of pyruvate to acetate and malate (Fig. 4). The possession of these pathways and the lack of a TCA cycle are similar features to those of saccharimonadial bins and support fermentative metabolism, as reported in previous studies (Wrighton et al., 2012; 2014; Albertsen et al., 2013; Danczak et al., 2017; Lemos et al., 2019; 2020; Starr et al., 2018; Sieber et al., 2019; Moreira et al., 2021; Yakimov et al., 2021). A moranbacterial bin (HHAS9) possessed chitinase (DEAMOMGP_00832 and DEAMOMGP_00955) (Fig. 4), but not the genes to convert N-acetylglucosamine to other compounds. The nomurabacterial bin (HHAS7) possessed the copper-containing nitrite reductase gene (JCNBLHJH_00228, nirK) (Fig. 4). Some members of Parcubacteria are known to be involved in nitrite reduction (Castelle et al., 2017; Danczak et al., 2017; He et al., 2021). In addition, several Chitinophagales bins (HHAS12 and HHAS13) possessed the nitric oxide reductase subunit B/C (norB/C) (CLEEFKKN_01663, CLEEFKKN_01664, AECFEMFL_01795, and AECFEMFL_01795) and nitrous-oxide reductase (nosZ) (CLEEFKKN_01654 and AECFEMFL_02605). Therefore, Nomurabacteria were partially responsible for denitrification along with Chitinophagaceae in the activated sludge process.
The gracilibacterial bin (HHAS10) had negligible central carbon metabolism. It possessed only pyruvate kinase (FNKGEGDK_00842), malate dehydrogenase (FNKGEGDK_00357), and 2-oxoglutarate/2-oxoacid ferredoxin oxidoreductase (FNKGEGDK_00164 and FNKGEGDK_00166). Although the poor metabolic potential of Gracilibacteria has also been reported (Sieber et al., 2019), the gracilibacterial genomes in previous studies were mainly reconstructed from other habitats, such as oral and ground water samples. The genome size of the HHAS10 bin was 1.3 Mbp, which is similar to that of other Gracilibacteria (Sieber et al., 2019), and completeness was relatively high (Table 1). Nevertheless, it was not possible to predict the metabolic potential of Gracilibacteria reconstructed in the present study using the current databases. The accumulation of genomic information on Gracilibacteria in activated sludge is necessary to construct substantial databases. In contrast, the gracilibacterial bin (HHAS10) possessed four copies of peptidase belonging to the M23 family (Table S4), which lyses the cell walls of other microorganisms. The HHAS10 bin also possessed a phospholipase gene (FNKGEGDK_00603, FNKGEGDK_00841), whereas Chitinophagales possessed an ABC transporter (MHHEHLFG_00269, MHHEHLFG_00294, MHHEHLFG_00428, MHHEHLFG_01772, MHHEHLFG_01773, MHHEHLFG_01879, KHEBLPDM_00308, KHEBLPDM_00309, and KHEBLPDM_02272), which releases phospholipids (Fig. 4). Therefore, the metabolic flow of phospholipids between Chitinophagales and Gracilibacteria in activated sludge was considerable. The HHAS10 bin possessed a homolog of polyhydroxyalkanoate (PHA) depolymerase (FNKGEGDK_00792), which showed >40% homology (<1e-65) to PHA depolymerases of known species (Table 2). This feature may help to obtain an energy source, even though Gracilibacteria have negligible central carbon metabolism. In general, PHA is degraded by PHA depolymerase to monomers, such as 3HB, which are then oxidized to acetoacetyl-CoA in a reaction catalyzed by 3HB dehydrogenase. This is then converted to acetyl-CoA by β-ketothiolase (Ong et al., 2017). Although Candidatus Parcunitrobacter nitroensis belonging to Parcubacteria also possessed PHB, which is a PHA, depolymerase, and peptidase that acts extracellularly and converts PHB to acetate, suggesting that PHB may be used as a carbon source (Castelle et al., 2017), no genes related to the reaction pathway of hydroxybutyrate in the HHAS10 bin were identified. Based on an amino acid sequence homology search using the NCBI-nr database, we found that the genome of HHAS10 bin had a surface layer protein (FNKGEGDK_00198) that was widely conserved in gracilibacterial genomes with high similarity (Table 2 and Fig. S4). Besides, the proteins showed 28% (41/145 bp, 3e-15) and 27–30% (<1e-16) homology with the PHB depolymerases of Candidatus Parcunitrobacter nitroensis (OWK27304.1) and other taxa (Myxococcales, Sorangium cellulosum, and Streptomyces sp.), respectively. Further studies are needed on the generality and roles of PHA/PHB depolymerases in Gracilibacteria.
Table 2.
Summary of genes related to polyhydroxyalkanoate and polyhydroxybutyrate depolymerases in the
Gracilibacteria bin HHAS10
| Locus tags |
Amino acid identity to known
proteins of representative taxonomies based on the NCBI-nr database |
| Description |
Accession no. |
Identities |
e-value |
| FNKGEGDK_00792 |
polyhydroxyalkanoate depolymerase [Alteromonas oceani] |
WP_123327345.1 |
138/317 (44%) |
6e-68 |
| polyhydroxyalkanoate depolymerase [Polynucleobacter paneuropaeus] |
WP_215313699.1 |
144/340 (42%) |
7e-68 |
| polyhydroxyalkanoate depolymerase [Alteromonas lipolytica] |
WP_070177576.1 |
137/317 (43%) |
2e-67 |
| polyhydroxyalkanoate depolymerase [Polynucleobacter wuianus] |
WP_216235107.1 |
144/340 (42%) |
2e-67 |
| polyhydroxyalkanoate depolymerase [Marisediminitalea aggregata] |
WP_073324272.1 |
137/317 (43%) |
4e-66 |
| FNKGEGDK_00198 |
hypothetical protein US76_00085 [Parcubacteria GW2011_GWA2_38_13b] |
OWK27304.1 |
41/145 (28%) |
3e-15 |
| S-layer homology domain-containing protein [Candidatus Gracilibacteria bacterium] |
MBP9812078.1 |
305/443 (69%) |
0 |
| S-layer homology domain-containing protein [Candidatus Gracilibacteria bacterium] |
MBC7498061.1 |
267/427 (63%) |
0 |
| Ricin and poly(3-hydroxybutyrate) depolymerase fusion [Myxococcales bacterium] |
MCA9656682.1 |
64/240 (27%) |
9e-22 |
| Ricin and poly(3-hydroxybutyrate) depolymerase fusion [Sorangium cellulosum] |
KYF79897.1 |
65/225 (29%) |
4e-19 |
| poly(3-hydroxybutyrate) depolymerase [Streptomyces sp. TLI_55] |
SNX55894.1 |
87/292 (30%) |
4e-17 |
Conclusions
In the present study, the metabolic potential of Patescibacteria was predicted from the MAGs of activated sludge samples, and the physiological role of Patescibacteria in activated sludge was estimated. The genomes of three Saccharimonadia, three Parcubacteria, and one Gracilibacteria species revealed a lack of de novo nucleotide synthesis, amino acid synthesis, phospholipid synthesis, and a full TCA cycle. Ten reconstructed genomes showed a strong positive correlation of relative abundance with Chitinophagales based on 16S rRNA genes. Metabolic interactions between a member of Saccharimonadia and Chitinophagales via N-acetylglucosamine, between a member of Parcubacteria and Chitinophagales via nitrogen compounds related to denitrification, and between Gracilibacteria and Chitinophagales via phospholipids in activated sludge were supported by metabolic predictions from 10 recovered Patescibacteria MAGs and five Chitinophagales MAGs. The high abundance of peptidases in Gracilibacteria suggests their role in cell lysis in activated sludge. Further studies related to visualization with FISH and the enrichment of Patescibacteria are necessary to elucidate the in situ physiological roles of Patescibacteria in the activated sludge process.
Citation
Fujii, N., Kuroda, K., Narihiro, T., Aoi, Y., Ozaki, N., Ohashi, A., and Kindaichi, T. (2022) Metabolic Potential of the Superphylum Patescibacteria Reconstructed from Activated Sludge Samples from a Municipal Wastewater Treatment Plant. Microbes Environ 37: ME22012.
https://doi.org/10.1264/jsme2.ME22012
Acknowledgements
This work was supported by JSPS KAKENHI, Grant Numbers JP16H04833, and JP20H02287.
References
- Albertsen, M., Hugenholtz, P., Skarshewski, A., Nielsen, K.L., Tyson, G.W., and Nielsen, P.H. (2013) Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol
31: 533–538.
- Anantharaman, K., Brown, C.T., Hug, L.A., Sharon, I., Castelle, C.J., Probst, A.J., et al. (2016) Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun
7: 13219.
- Asnicar, F., Thomas, A.M., Beghini, F., Mengoni, C., Manara, S., Manghi, P., et al. (2020) Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun
11: 2500.
- Awata, T., Goto, Y., Kuratsuka, H., Aoi, Y., Ozaki, N., Ohashi, A., et al. (2021) Reactor performance and microbial community structure of single-stage partial nitritation anammox membrane bioreactors inoculated with Brocadia and Scalindua enrichment cultures. Biochem Eng J
170: 107991.
- Bankevich, A., Nurk, S., Antipov, D., Gurevich, A.A., Dvorkin, M., Kulikov, A.S., et al. (2012) SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol
19: 455–477.
- Bolger, A.M., Lohse, M., and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics
30: 2114–2120.
- Bolyen, E., Rideout, J.R., Dillon, M.R., Bokulich, N.A., Abnet, C.C., Al-Ghalith, G.A., et al. (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol
37: 852–857.
- Brown, C.T., Hug, L.A., Thomas, B.C., Sharon, I., Castelle, C.J., Singh, A., et al. (2015) Unusual biology across a group comprising more than 15% of domain Bacteria. Nature
523: 208–211.
- Castelle, C.J., Brown, C.T., Thomas, B.C., Williams, K.H., and Banfield, J.F. (2017) Unusual respiratory capacity and nitrogen metabolism in a Parcubacterium (OD1) of the Candidate Phyla Radiation. Sci Rep
7: 40101.
- Castelle, C.J., Brown, C.T., Anantharaman, K., Probst, A.J., Huang, R.H., and Banfield, J.F. (2018) Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat Rev Microbiol
16: 629–645.
- Chaudhari, N.M., Overholt, W.A., Figueroa-Gonzalez, P.A., Taubert, M., Bornemann, T.L.V., Probst, A.J., et al. (2021) The economical lifestyle of CPR bacteria in groundwater allows little preference for environmental drivers. Environ Microbiome
16: 24.
- Chen, I., and Gotschlich, E.C. (2001) ComE, a competence protein from Neisseria gonorrhoeae with DNA-Binding Activity. J Bacteriol
183: 3160–3168.
- Danczak, R.E., Johnston, M.D., Kenah, C., Slattery, M., Wrighton, K.C., and Wilkins, M.J. (2017) Members of the candidate phyla radiation are functionally differentiated by carbon and nitrogen-cycling capabilities. Microbiome
5: 112.
- Dinh, H.T.T., Kambara, H., Harada, Y., Matsushita, S., Aoi, Y., Kindaichi, T., et al. (2021) Bioelectrical methane production with an ammonium oxidative reaction under the no organic substance condition. Microbes Environ
36: ME21007.
- Dudek, N.K., Sun, C.L., Burstein, D., Kantor, R.S., Goltsman, D.S.A., Bik, E.M., et al. (2017) Novel microbial diversity and functional potential in the marine mammal oral microbiome. Curr Biol
27: 3752–3762.
- Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics
28: 3150–3152.
- Graham, E.D., Heidelberg, J.F., and Tully, B.J. (2018) Potential for primary productivity in a globally-distributed bacterial phototroph. ISME J
12: 1861–1866.
- Hammer, Ø., Harper, D.A.T. and Ryan, P.D. (2001) PAST: Paleontological statistics software package for education and data analysis. Palaeontol Electronica
4: 9pp.
- Hanke, A., Hamann, E., Sharma, R., Geelhoed, J.S., Hargesheimer, T., Kraft, B., et al. (2014) Recoding of the stop codon UGA to glycine by a BD1-5/SN-2 bacterium and niche partitioning between Alpha- and Gammaproteobacteria in a tidal sediment microbial community naturally selected in a laboratory chemostat. Front Microbiol
5: 231.
- He, C., Keren, R., Whittaker, M.L., Farag, I.F., Doudna, J.A., Cate, J.H.D., et al. (2021) Genome-resolved metagenomics reveals site-specific diversity of episymbiotic CPR bacteria and DPANN archaea in groundwater ecosystems. Nat Microbiol
6: 354–365.
- He, X.S., McLean, J.S., Edlund, A., Yooseph, S., Hall, A.P., and Liu, S.Y. (2015) Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc Natl Acad Sci U S A
112: 244–249.
- Hosokawa, S., Kuroda, K., Narihiro, T., Aoi, Y., Ozaki, N., Ohashi, A., et al. (2021) Cometabolism of the superphylum Patescibacteria with anammox bacteria in a long-term freshwater anammox column reactor. Water (Basel, Switz)
13: 208.
- Hug, L.A., Baker, B.J., Anantharaman, K., Brown, C.T., Probst, A.J., Castelle, C.J., et al. (2016) A new view of the tree of life. Nat Microbiol
1: 16048.
- Hugenholtz, P., Tyson, G.W., Webb, R.I., Wagner, A.M., and Blackall, L.L. (2001) Investigation of candidate division TM7, a recently recognized major lineage of the domain bacteria with no known pure-culture representatives. Appl Environ Microbiol
67: 411–419.
- Ibrahim, A., Maatouk, M., Rajaonison, A., Zgheib, R., Haddad, G., Khalil, J.B., et al. (2021) Adapted protocol for Saccharibacteria cocultivation: two new members join the club of candidate phyla radiation. Microbiol Spectrum
9: e01069-21.
- Kambara, H., Shinno, T., Matsuura, N., Matsushita, S., Aoi, Y., Kindaichi, T., et al. (2022) Environmental factors affecting the community of methane-oxidizing bacteria. Microbes Environ
37: ME21074.
- Kanehisa, M., Sato, Y., and Morishima, K. (2016) BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol
428: 726–731.
- Kang, D.D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019) MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ
7: e7359.
- Kantor, R.S., Wrighton, K.C., Handley, K.M., Sharon, I., Hug, L.A., Castelle, C.J., et al. (2013) Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. mBio
4: e00708-13.
- Katoh, K., and Standley, D.M. (2013) MAFFT Multiple Sequence Alignment Software Version 7: improvements in performance and usability. Mol Biol Evol
30: 772–780.
- Kindaichi, T., Nierychlo, M., Kragelund., C., Nielsen, J.L., and Nielsen, P.H. (2013) High and stable substrate specificities of microorganisms in enhanced biological phosphorus removal plants. Environ Microbiol
15: 1821–1831.
- Kindaichi, T., Yamaoka, S., Uehara, R., Ozaki, N., Ohashi, A., Albertsen, M., et al. (2016) Phylogenetic diversity and ecophysiology of Candidate phylum Saccharibacteria in activated sludge. FEMS Microbiol Ecol
92: fiw078.
- Lemos, L.N., Medeiros, J.D., Dini-Andreote, F., Fernandes, G.R., Varani, A.M., Oliveira, G., et al. (2019) Genomic signatures and co-occurrence patterns of the ultra-small Saccharimonadia (phylum CPR/Patescibacteria) suggest a symbiotic lifestyle. Mol Ecol
28: 4259–4271.
- Lemos, L.N., Manoharan, L., Mendes, L.W., Venturini, A.M., Pylro, V.S., and Tsai, S.M. (2020) Metagenome assembled-genomes reveal similar functional profiles of CPR/Patescibacteria phyla in soils. Environ Microbiol Rep
12: 651–655.
- Mielczarek, A.T., Kragelund, C., Eriksen, P.S., and Nielsen, P.H. (2012) Population dynamics of filamentous bacteria in Danish wastewater treatment plants with nutrient removal. Water Res
46: 3781–3795.
- Minh, B.Q., Schmidt, H.A., Chernomor, O., Schrempf, D., Woodhams, M.D., Von H.A., et al. (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol
37: 1530–1534.
- Moreira, D., Zivanovic, Y., López-Archilla, A.I., Iniesto, M., and López-Garcia, P. (2021) Reductive evolution and unique predatory mode in the CPR bacterium Vampirococcus lugosii. Nat Commun
12: 2454.
- Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A.C., and Kanehisa, M. (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res
35: 182–185.
- Nakai, R. (2020) Size matters: Ultra-small and filterable microorganisms in the environment. Microbes Environ
35: ME20025.
- Nelson, W.C., and Stegen, J.C. (2015) The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Front Microbiol
6: 713.
- Nielsen, P.H., Mielczarek, A.T., Kragelund, C., Nielsen, J.L., Saunders, A.M., and Kong, Y. (2010) A conceptual ecosystem model of microbial communities in enhanced biological phosphorus removal plants. Water Res
44: 5070–5088.
- Ong, S.Y., Chee, J.Y., and Sudesh, K. (2017) Degradation of polyhydroxyalkanoate (PHA): a review. J Sib Fed Univ Biol
10: 211–225.
- Parks, D.H., Imelfort, M., Skennerton, C.T., Hugenholtz, P., and Tyson, G.W. (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res
25: 1043–1055.
- Phan, H.V., Kurisu, F., Kiba, K., and Furumai, H. (2021) Optimized cultivation and syntrophic relationship of anaerobic benzene-degrading enrichment cultures under methanogenic conditions. Microbes Environ
36: ME21028.
- Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res
41: D590–D596.
- Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N.N., Anderson, I.J., Cheng, J.F., et al. (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature
499: 431–437.
- Seemann, T. (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics
30: 2068–2069.
- Shaffer, M., Borton, M.A., McGivern, B.B., Zayed, A.A. La Rosa, S.L., Solden, L.M., et al. (2020) DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res
48: 8883–8900.
- Sieber, C.M.K., Paul, B.G., Castelle, C.J., Hu, P., Tringe, S.G., Valentine, D.L., et al. (2019) Unusual metabolism and hypervariation in the genome of a Gracilibacterium (BD1-5) from an oil-degrading community. mBio
10: e02128-19.
- Singleton, C.M., Petriglieri, F., Kristensen, J.M., Kirkegaard, R.H., Michaelsen, T.Y., Andersen, M.H., et al. (2021) Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing. Nat Commun
12: 2009.
- Soro, V., Dutton, L.C., Sprague, S.V., Nobbs, A.H., Ireland A.J., Sandy, J.R., et al. (2014) Axenic culture of a Candidate division TM7 bacterium from the human oral cavity and biofilm interactions with other oral bacteria. Appl Environ Microbiol
80: 6480–6489.
- Starr, E.P., Shi, S., Blazewicz, S.J., Probst, A.J., Herman, D.J., Firestone, M.K., et al. (2018) Stable isotope informed genome-resolved metagenomics reveals that Saccharibacteria utilize microbially-processed plant-derived carbon. Microbiome
6: 122.
- Takebe, H., Tominaga, K., Fujiwara, K., Yamamoto, K., and Yoshida, T. (2020) Differential responses of a coastal prokaryotic community to phytoplanktonic organic matter derived from cellular components and exudates. Microbes Environ
35: ME20033.
- Wrighton, K.C., Thomas, B.C., Sharon, I., Miller, C.S., Castelle, C.J., VerBerkmoes, N.C., et al. (2012) Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science
337: 1661–1665.
- Wrighton, K.C., Castelle, C.J., Wilkins, M.J., Hug, L.A., Sharon, I., Thomas, B.C., et al. (2014) Metabolic interdependencies between phylogenetically novel fermenters and respiratory organisms in an unconfined aquifer. ISME J
8: 1452–1463.
- Yakimov, M.M., Merkel, A.Y., Gaisin, V.A., Pilhofer, M., Messina, E., Hallsworth, J.E., et al. (2021) Cultivation of a vampire: ‘Candidatus Absconditicoccus praedator’. Environ Microbiol
24: 30–49.
- Zhang, T., Shao, M.F., and Ye, L. (2012) 454 Pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J
6: 1137–1147.