Review
Angiogenic defects in preeclampsia: What is known, and how are such defects relevant to preeclampsia pathogenesis?
2013 Volume 1 Issue 2 Pages 57-65
Details
2013 Volume 1 Issue 2 Pages 57-65
Preeclampsia is a devastating pregnancy-associated hypertensive syndrome. Although it is quite common, the pathophysiology of preeclampsia is not yet clear and remains “a disease of theory”. Angiogenic defect hypotheses have been intensively investigated, and some biomarkers have been independently analyzed as pathogenic clinical target molecules without direct proof of their roles in preeclampsia. In this review, we assessed an up-to-date list of proposed angiogenic defects for their relevance to preeclampsia. In addition, we introduce our working hypothesis of preeclampsia pathophysiology, which involves interactions between metabolic and angiogenic defects.
Preeclampsia is a devastating syndrome characterized by pregnancy-specific new-onset hypertension and proteinuria. The overall incidence is approximately 5–8% of all pregnant women.1) Presenting symptoms can be highly variable, starting from seemingly innocuous complaints, such as heartburn or headache, and eventually progressing to multi-systemic, life-threatening dysfunctions, such as HELLP (hemolysis, elevated liver function tests, low platelets) syndrome and eclampsia (characterized by seizures).1) Therapeutic options are limited at present; only delivering the baby provides an absolute ‘cure’ for worsening maternal symptoms. The preeclampsia-associated newborn is often premature, resulting in significant perinatal morbidity and mortality, as well as effects on future development. It is estimated that approximately 15% of preterm births are due to preeclampsia.
Despite its fairly high incidence,1) our understanding of preeclampsia pathophysiology is limited. Preeclampsia has been recognized as “a disease of theories” because several hypotheses have been advanced to explain its pathogenesis. Among these hypotheses, the angiogenic molecular defects theory has received considerable attention, with some pilot studies initiated to target these defects as pathogenic molecules. Most of the angiogenic defects theory is based on clinical molecular marker observations using cDNA-carrying viral vectors to assess overexpression of foreign proteins. In this review, we analyze whether the angiogenic molecular defects theory is pathophysiologically supported by enough clinical/biological evidence to explain preeclampsia. We also introduce a potential interaction between catechol-metabolizing defects and angiogenic regulation deficiency in preeclampsia pathogenesis.
It is widely accepted that placental hypoxia can trigger preeclampsia by initiating a sequence of events that ultimately results in the maternal symptoms of hypertension and proteinuria.1) Placental hypoxia can be caused by diverse factors, such as maternal hypoxia, maternal hypotension, and placental ischemia, but in the context of preeclampsia, it is considered secondary to defective placental vasculature.2)
The placenta is an essential organ for exchanging waste and nutrients between mother and fetus. An embryonic cell known as the cytotrophoblast is essential for proper development of the placenta and its vasculature. One cytotrophoblast derivative, the extravillous cytotrophoblast, invades the uterine matrix, makes direct contact with the maternal blood supply, and subsequently replaces the smooth muscle cell layer of the maternal vasculature.1) This remodeling via trophoblast invasion results in vessels that are maternal-derived and vasoactive substance-resistant. Because the vessels are independent from the influence of maternal blood pressure regulation, they tend to be dilated with low resistance.1) Preeclampsia is characterized by shallow trophoblast invasion. As a result, placental circulation does not provide sufficient blood supply to meet embryonic demand due to high resistance, non-dilated vessels that reflect inadequate remodeling of maternal spiral arteries.1)
How does placental hypoxia cause the maternal symptoms of preeclampsia? This important question is still openly debated. One theory is that reduced placental perfusion results in the production of numerous placenta-derived circulatory agents, which cause the maternal symptoms.
Much attention has been given to the topic of angiogenic imbalance in preeclampsia pathogenesis. However, this theory is rather complicated due to the biology of vascular endothelial growth factor (VEGF) (Figure 1).

VEGF binding partners and their effects on VEGF activity.
Vascular endothelial growth factor (VEGF) can bind to several molecules, and certain interactions do not detectably alter VEGF activity, as assessed by a commercially available ELISA assay.
Angiogenic abnormalities in preeclampsia were first reported by Selvaggi et al., who used the chick embryo chorioallantoic membrane angiogenic assay to demonstrate that decidua in preeclamptic women exhibited higher angiogenic activity than in a normal pregnancy.3,4) Although the angiogenesis imbalance theory is popular, these important findings have not been described well in the literature. Such an enhanced angiogenesis signal could likely be a compensatory reaction to shallow extravillous trophoblast invasion and associated perfusion defects and placental hypoxia.3,4) Selgaggi’s findings were important because they revealed that hypoxic placental decidua in preeclampsia exhibited enhanced angiogenic activity.
Despite this evidence of enhanced angiogenic activity in the preeclamptic placenta, preeclampsia has been considered an anti-angiogenic state,5,6,7,8) although this has not been proven yet. This theoretical shift in preeclampsia pathogenesis, from ‘angiogenic’ to ‘anti-angiogenic’, is linked to the implication of neutralizing soluble VEGF receptors. Soluble VEGF type1 receptor (also known as soluble Fms-like tyrosine kinase-1 [sFlt-1]) has been hypothesized as an important candidate molecule associated with preeclampsia pathogenesis.9,10,11,12) In 1998, Clark et al. discovered that serum from pregnant women contained a VEGF-binding protein not present in serum from men or non-pregnant women.13) Vuorela et al. initially reported that VEGF was bound to soluble proteins in amniotic fluid and maternal serum,9) later discovering in 2000 that sFlt-1 was significantly elevated in the amniotic fluid of women with preeclampsia.10) This work was the seminal report implicating sFlt-1 in preeclampsia. Fisher et al. later demonstrated that cytotrophoblasts from preeclamptic placentas exhibited higher levels of sFlt-1 in vitro compared to control cytotrophoblasts from normal placentas.12) In 2003, Sugimoto et al. discovered that injecting sFlt-1 protein or anti-VEGF antibodies into mice resulted in proteinuria, likely via neutralization of the bioavailable form of VEGF.14) Later in the same year, Maynard et al. reported that the concentration of sFlt-1 in serum from preeclamptic women was higher compared to women with normal pregnancies and that viral vector-mediated delivery of human sFlt-1 cDNA caused endotheliosis and hypertension in male, non-pregnant female and pregnant female rats.15) Their study also demonstrated that serum from preeclamptic women was anti-angiogenic compared to normal control pregnancies when analyzed by the endothelial tube-formation assay. Notably, incubation with both 10 ng/ml VEGF (a concentration approximately 1,000×higher than normal pregnancy) and 10 ng/ml placental growth factor (PlGF) (a concentration approximately 25×higher than normal pregnancy) abolished this anti-angiogenic property of preeclamptic serum.15)
As described above, angiogenic and anti-angiogenic imbalance theories have been intensively investigated. However, the important fundamental question that remains is whether VEGF biological activity is high or low in preeclampsia.
The study described above by Maynard et al., showing that adenoviral-mediated sFlt-1 overexpression resulted in preeclampsia-like symptoms in rats,15) led to the widespread but untested concept that sFlt-1 may play a causal role in preeclampsia pathogenesis. Maynard’s theory is mainly based on the anti-angiogenic hypothesis of preeclampsia; however, many fundamental issues need to be addressed in this model. First, the concentration achieved by adenoviral-mediated overexpression of human sFlt-1 is approximately 215.5 ng/ml in rats, which is significantly higher compared to that observed in severe preeclampsia (7.6 ng/ml).15) The authors also described a lower concentration of human sFlt-1 (7.3±3.2 ng/ml, also produced by adenoviral vector) that caused a milder preeclampsia phenotype in rats, although it is not clear whether these experiments were performed in pregnant, male, or female rats.15) The second question is the relevance of sFlt-1 species and isoforms used in these studies. It is unknown which sFlt-1 isoforms increase with preeclampsia, a highly important point when producing appropriate animal models. For example, the primate-specific variant sFlt-1-14 has been reported in preeclampsia16) but not tested as a preeclampsia candidate molecule in animal models. Sugimoto et al. employed a human sFlt-1/human IgGFc chimera that was intravenously injected into mice.14) Maynard’s work and subsequent adenovirus-based preeclampsia rat models used mouse sFlt-1 (lacking 31 amino acid carboxyl residues) and immunoglobulin-like loop 4,15) which is essential for receptor dimerization of the extracellular domains of sFlt-1.17,18) To solve these problems, Suzuki et al. used an adenovirus to express full-length mouse sFlt-1 in a mouse model.19) They found that only a very high concentration of adenovirus vector (2×109, compared to 1×108 in Maynard’s work) produced a preeclampsia-like phenotype with 100 ng/ml of plasma sFlt-1 in mice; however, a concentration of 1×109 could induce a similar concentration of plasma sFlt-1 in mice and cause gestational proteinuria without gestational hypertension.19) Moreover, the hypertension observed in this model was attenuated by mouse VEGF or PlGF adenovirus administration, but proteinuria was not.19) Adenovirus infection results in serious liver damage,20,21) which could not be ignored as a confounding factor in all adenoviral preeclampsia models. In this regard, Kumasawa et al. established an elegant preeclampsia mouse model by transducing a lentiviral vector expressing human sFlt-1 into zona pellucida-free blastocysts to establish pregnancy placenta-specific sFlt-1 expression in mice.22) By overexpressing human sFlt-1, their model somewhat mimicked pregnancy induced hypertension with proteinuria.22) Hypertension was rescued by administration of the cholesterol-lowering agent pravastatin, likely associated with elevation of PlGF levels.22) Pravastatin showed a trend of attenuated proteinuria in their model.22)
The assumption was that so-called ‘free’ VEGF, which can be measured with enzyme-linked immunosorbent assay (ELISA), is the only bioactive form of VEGF; however, this has never been biologically proven, and recent evidence has challenged this view.23) High VEGF levels have been reported for preeclampsia in several biological fluids, including plasma, serum and amniotic fluid, by radioimmunoassay (RIA) or capture enzyme immunoassay.23) However, commercially available VEGF ELISA kits revealed suppressed VEGF levels in preeclampsia compared to normal pregnancy.23) This discrepancy in measurements with different methodologies led researchers to speculate that VEGF-binding molecules interfere with antibody binding in ELISAs, which may measure unbound free VEGF levels.15,23) Whether free VEGF is the only active form of VEGF has not been experimentally proven. When estimated using RIA, the VEGF concentration was more than 20×higher than that of sFlt-1, even in preeclampsia; when estimated using ELISA, VEGF levels were much lower than those of sFlt-1, even in normal pregnancy.23) Therefore, although it is true that sFlt-1 levels are higher in most cases of preeclampsia,5,15,24) sFlt-1 levels cannot completely explain the reduced VEGF levels, as observed by ELISA; rather, it must be due to other molecules that bind VEGF without affecting its biological activity. Some reports have indicated that VEGF activity is high in preeclampsia.25,26,27) One molecule that may bind VEGF is α2-macroglobulin, and this interaction does not influence VEGF-induced receptor activation.28,29) Moreover, alternative splice variants, such as VEGF165b, act as weak agonists for the angiogenic receptor VEGF receptor 2. VEGF165b inhibits VEGF165-induced intracellular signaling and thus plays an antagonistic role in VEGF165-mediated angiogenesis.23,25,30) Nevertheless, the ‘biological activity of VEGF’ is often confused with the ‘unbound form of VEGF’ in the literature. This point needs to be carefully investigated in future research (Figure 1).
Pregnant women who develop preeclampsia often exhibit high sFlt-1 levels with low free VEGF (but not total VEGF), as measured by ELISA, and low PlGF. Drug administration in pregnant women is challenging due to potential teratogenicity and toxicity. Therefore, the ideal preeclampsia therapy would remove some pathogenic factors from circulatory blood without administering a drug. Thadhani et al. developed a unique way to test the anti-angiogenic theory of preeclampsia.31) sFlt-1 is a positively charged circulatory protein, and they focused on this characteristic as a therapeutic target.31) Based on the unproven sFlt-1 theory, they tried to eliminate positively-charged sFlt-1 from preeclamptic serum using an extracorporeal affinity adsorption column,31) specifically, clinically-available negatively-charged dextran sulfate columns (DSCs) which would theoretically absorb positively-charged sFlt-1. In their analysis, DSCs decreased sFlt-1 levels in the plasma of preeclamptic women from 13,471±3,358 pg/ml to 10,825±2,918 pg/ml (n=5, 20.1±5.1% reduction; mean±s.d.).31) This decrease in sFlt-1 was associated with a 56.8±6.7% reduction in the urine protein-creatinine ratio (3,326±2,306 to 1,349±811).31) Although an interesting idea, this pilot study did not provide information about how the system works. sFlt-1 is not the only positively-charged molecule with a potential pathogenic role in preeclampsia. Fibrinogen is also positively-charged and highly relevant to blood rheology and coagulation, and DSC apheresis decreases its content by 18.0±11.3%.31) Low density lipoprotein cholesterol, other inflammatory proteins, and immunoglobulins (especially angiotensin receptor autoantibodies) could also be removed from preeclamptic maternal serum by DSCs. Performing apheresis itself may alter blood pressure and rheology. Wang et al. elegantly demonstrated that the pregnancies of 9 very preterm preeclamptic women were prolonged 3 to 49 additional days by utilizing apheresis with heparin-mediated extracorporeal low-density lipoprotein precipitation (H.E.L.P.) instead of DSCs.32) H.E.L.P. apheresis in preeclamptic women was associated with reductions in tumor necrosis factor-α, soluble vascular cell adhesion molecule 1, E-selectin, endothelin 1, lipopolysaccharide-binding protein, homocysteine, fibrinogen and C-reactive protein.32) H.E.L.P. also reduced apolipoprotein B–containing lipoproteins by almost 50% in preeclamptic women.32) H.E.L.P. apheresis is likely not associated with the reduction in sFlt-1 because, as Thadhani et al. stated, it is less effective than DSCs in reducing sFlt-1.31) DSC apheresis reduces lipoprotein levels; therefore, interpreting the DSC data described above is more complicated than simple sFlt-1 removal.
Catechol-O-methyltransferase (Comt) is a candidate gene in various psychiatric disorders, such as schizophrenia, that encodes a catabolic enzyme fundamental in the metabolism of diverse catechols, including catecholamines and catecholestrogens. Estradiol is metabolized by cytochrome p450, and synthesized 17-hydroxyestradiol (a catecholestrogen) is a substrate for COMT. This catecholestradiol is then metabolized by COMT into 2-methoxyestradiol (2-ME), in a rate-limiting step. 2-ME inhibits hypoxia-inducible factor-1 α (HIF-1α), possibly by destabilizing microtubules,33) allowing it to act as an anti-angiogenic molecule in a context-dependent manner. Clinical evaluations are currently underway to investigate 2-ME as a new oral therapeutic agent in the treatment of cancer.34,35,36) Normal pregnant women exhibit increased concentrations of 2-ME that peak at term (from 2–15 nM in the 11–16th week of pregnancy to 18–96.21 nM in the 37–40th week of pregnancy).37)
Barnea et al. first reported the suppression of placental COMT activity in preeclampsia in 1988,38) but its significance was not mechanistically analyzed. We recently demonstrated that deficiencies in COMT and 2-ME are associated with placental hypoxia and preeclampsia-like symptoms in mice.33) Comt-deficient mice (Comt-/-) display pregnancy induced hypertension with proteinuria due to the absence of 2-ME, and administration of exogenous 2-ME (10 ng subcutaneous injection from day 10 of pregnancy) ameliorates hypertension, proteinuria, placental defects, acute atherosis and glomerular and placental endothelial damage in pregnant Comt-/- mice (Figure 2). The preeclampsia-like phenotypes of pregnant Comt-/- mice were largely corroborated by Stanley et al.,39) and any differences could be explained by the mouse strain used (SV129 in our study, C57BL/6 in the study by Stanley et al.).33)
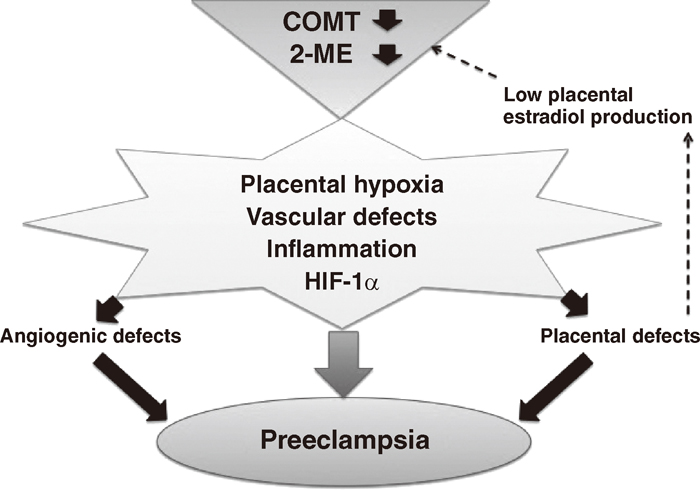
Working hypothesis of COMT deficiency and the pathogenesis of preeclampsia.
Deficiency in catechol-O-methyltransferase (COMT) and the associated suppression of 2-methoxyestradiol (2-ME) levels may result in hypoxia, vascular defects, hypoxia-inducible factor-1 alpha accumulation and inflammation in placentas. These defects in placental homeostasis induce defects in both angiogenesis and the placenta. Placental defects lead to suppression of estradiol production from placentas, resulting in further suppression of 2-ME. This vicious cycle leads to preeclampsia.
The underlying mechanisms of COMT and 2-ME deficiency in the pathogenesis of a preeclampsia-like phenotype may be linked to HIF-1α accumulation in Comt-/- mice placentas (Figure 2), which is associated with an enhanced inflammatory response and endothelial damage. Suppression of hypoxia-induced HIF-1α accumulation by 2-ME is correlated with sFlt-1 suppression. 2-ME decreases NK cell accumulation and interferon-γ production, ameliorating endothelial damage in Comt-/- mice (Figure 2). 2-ME also induces trophoblast invasion under hypoxic conditions by suppressing HIF-1α and transforming growth factor-beta 3, likely via trophoblast-to-endothelial-like cell conversion, suggesting that 2-ME is a fundamental player in maintaining placental homeostasis40) and supporting the theory proposed by Caniggia et al.41) 2-ME may directly function as a vasodilator and inhibit vasospasm42,43,44,45) in pregnant women.
COMT enzymatic activity exhibits a trimodal frequency distribution in human populations, which could be explained by the presence of a functional polymorphism in the coding sequence.46) One such polymorphism, rs4680 (G-A nucleotide substitution), results in a missense mutation of Val to Met at position COMT158.46) Human COMTMet158 has a lower stability with a lower enzymatic activity at physiological temperatures,46) and approximately 25–30% of humans possess this polymorphism. Sata et al. reported that this functional Comt polymorphism is associated with fetal growth restriction.47) In addition to COMT,Met158 Nackley et al. reported that haplotypes in multiple single nucleotide polymorphisms (SNPs) of the Comt gene affect COMT mRNA stability,48) suggesting that preeclampsia susceptibility is associated with genomic alterations. It must be emphasized that these hypotheses have yet to be investigated, and other SNP analyses should be conducted in multiple families in different population cohorts. COMT deficiency cannot explain all phenotypical preeclampsia variants in humans. Preeclampsia is likely a heterogeneous collection of diverse pathomechanisms, and COMT deficiency may only play a role in select patient populations. For example, maternal COMTMet158 has been associated with preeclampsia in Korean and Chinese cohorts,49,50) and a Norwegian population cohort (HUNT2) study revealed that the maternal haplotype for low COMT activity was associated with recurrent preeclampsia.51) COMT activity may be driven by factors other than SNPs, such as translational control or other diverse pathways that lead to decreased activity.52,53)
Elevated homocysteine levels may be associated with an increased risk of preeclampsia.54,55) The mechanisms by which homocysteine contributes to preeclampsia pathogenesis could be explained by thrombosis of placental vasculature, inhibition of nitric oxide production and general oxidative stress.56,57,58,59) At normal concentrations, homocysteine primarily proceeds along pathways associated with less reactive oxidative methionine or cysteine; however, excess homocysteine is increasingly converted to S-adenosyl homocysteine (SAH), which is a potent endogenous COMT inhibitor (Figure 3).60) If SAH accumulates in sufficient concentrations to inhibit COMT activity, O-methylation and inactivation of circulatory endogenous catecholamines and other catechols decrease.60) An increase in circulatory catecholamines can result in sympathetic nervous system overactivation, leading to an increased risk of cardiovascular morbidity and mortality.60)

Proposed hypothetical models in the interaction of hyperhomocysteinemia and preeclampsia onset via COMT deficiency.
Due to deficiencies in nutrition and activities of diverse enzymes, homocysteine levels are elevated. Homocysteine is converted into s-adenosyl-homocysteine, an endogenous inhibitor of the catechol-O-methyltransferase (COMT) enzyme. Therefore, women (or a combination of the placenta and pregnant mother) with SNPs associated with low COMT enzymatic activity can exhibit severely suppressed COMT activity, leading to preeclampsia.
Men with high serum homocysteine levels who are also homozygous for the low-activity Comt gene (Val158Met isoform; COMTLL) are at increased risk of coronary heart disease.61) This finding may be relevant to the pathogenesis of preeclampsia in women who are homozygous for COMTLL. The possibility that homocysteine-induced preeclampsia is dependent on Comt genotype could be why hyperhomocysteinemia did not lead to preeclampsia in previous animal studies with wild-type rodents.62,63) It is also possible that placental, but not maternal, or both Comt genotypes are much more relevant to interactions with hyperhomocysteinemia.
The pathological interaction between hyperhomocysteinemia and low COMT activity provides several potential pathomechanisms to explain why ComtLL women or women with other haplotypes associated with the low-activity COMT enzyme might be susceptible to preeclampsia. Increased levels of homocysteine (and SAH) could suppress COMT enzymatic activity and subsequently reduce 2-ME production (Figure 3). Similar to the findings in ComtLL men, hyperhomocysteinemia may also contribute to the defective metabolism of endogenous catecholamine levels in ComtLL women or women with the other haplotypes. Furthermore, deficiencies in potent homocysteine suppressors, such as B vitamins and folic acid, could increase the risk of preeclampsia.64,65,66,67) Folic acid supplementation in the second trimester of pregnancy has been shown to significantly reduce both serum homocysteine levels and the risk of preeclampsia.68)
More research is required to clarify these results, as the effects of folic acid therapy observed in this study could have been related to the Comt phenotype. These results could also pave the way to future studies that investigate the interaction of Comt SNPs and other genetic defects in the folic acid metabolism pathway, such as the methylenetetrahydrofolate reductase (Mthfr) C677T mutation,69) with the development of preeclampsia. In this regard, both maternal and fetal Comt haplotypes may be associated with a fetal Mthfr C677T mutation in preeclampsia pathogenesis.70)
Placentas might exhibit an anti-angiogenic state with compensatory induction of angiogenic signaling; however, it is not clear whether systemically preeclamptic women develop this state. Most evidence supporting the anti-angiogenesis state hypothesis is based on biomarker analysis rather than a mechanistically proven theory.5,6,7,8) Because virus-mediated overexpression models may lead us to overestimate preeclampsia pathogenesis,15,19,22,71,72,73,74) it would be extremely useful to have a relevant genetic mouse model. Such models may be phenotypically weak compared to those generated by adenovirus transfer33); however, weak but relevant phenotypes are common in genetic disease models. Despite similarities in general physiology, mouse disease models are not exact phenocopies of human pathophysiology, and achieving a ‘complete copy’ is impossible. This limitation has been well documented in genetic cancer models, in which phenotypes between mice and men overlap, but the tumor progression rate and associated outcomes can differ.
We must understand how to design proper, useful animal models, and we should not make misinterpretations based on the limitations of disease models. It is important to understand the limitation of each animal model, even if stronger phenotypes can be obtained via physiologically irrelevant interventions. Angiogenic abnormalities and the anti-angiogenic state in preeclampsia remain ‘unproven hypotheses.’ The differences between molecular biomarker analyses must therefore be carefully interpreted to understand the role of such biomarkers in disease pathogenesis.
This review focused on the angiogenic abnormality theory. Other highly relevant theories such as inflammation75,76) and angiotensin receptor agonistic autoantibodies77) have also been suggested as pathomechanisms of preeclampsia. Additionally, none of the animal preeclampsia models exhibit metabolic defects such as insulin resistance, which is an important associated condition in human preeclampsia. Future research is needed to identify mechanisms to understand preeclampsia pathogenesis and establish relevant animal models.
The authors declare no conflict of interest in this work. The authors’ laboratories are currently supported by grants from the Japan Society for the Promotion of Science to M.K. (24790329) and K.K. (23790381). K.K. is also supported by a Grant for Promoted Research (S2011-1, S2012-5) from Kanazawa Medical University and several foundation grants, including the Japan Research Foundation for Clinical Pharmacology, the Daiichi-Sankyo Foundation of Life Science, the Ono Medical Research Foundation, the NOVARTIS Foundation (Japan) for the Promotion of Science, the Takeda Science Foundation and the Banyu Life Science Foundation.