Abstract
Selenoglutathione (GSeH) is a water-soluble tripeptide, in which the sulfur atom of biologically important reductant glutathione (GSH) is replaced by a selenium atom, and exhibits a higher reducing activity than GSH. In this review, we overview the research on this selenium analogue of glutathione and look ahead to future research directions. Both solid-phase and liquid-phase methods can be used to chemically synthesize selenoglutathione diselenide (GSeSeG), the oxidized form of GSeH. Lesser amounts of GSeH are also found in the metabolic products of yeast and certain plants (garlic, sunflower sprouts, etc.) grown in a high-selenium medium. In the meantime, a biological method for the synthesis of GSeH using mutated yeast has recently been reported. Various applications of selenoglutathione in the field of biochemistry have already been explored. It has been reported that GSeSeG is an efficient catalyst for the oxidative folding of proteins. GSeSeG is also an excellent radical scavenger and potential detoxifier of intracellular xenobiotics. Recently, it has also been reported that GSeSeG has stress-reducing and antibacterial properties. Because GSeSeG has low toxicity, its unique reactivity is expected to be widely applied in the fields of applied biology and medicine.
1. Introduction
Selenium is an essential micronutrient, usually present as selenocysteine (Sec) in the body and contained in the reaction center of various enzymes [1,2]. Sec is a natural amino acid, in which the sulfur atom of cysteine (Cys) is replaced by a selenium atom, and is known as the 21st proteinogenic amino acid [3]. Research on the synthesis and function of peptides containing Sec is being actively conducted [4]. The chemical synthesis of artificial proteins, in which Cys has been mutated to Sec, and their unique biological functions are also attracting large interest of researchers [5–8]. In recent decades, the biological functions of selenouracil derivatives, which were found in nucleic acids, have also been actively studied [9,10]. The progress of these research reveals the potential of these selenium analogs of biomolecules as possible candidates of drug discovery [8].
Glutathione (GSH) is a water-soluble tripeptide with a molecular structure of γ-Glu-Cys-Gly, which is abundant in cells and plays important roles in maintaining redox homeostasis in the body and relieving stress through various redox reactions, such as decomposing free radicals and reactive oxygen species (ROS) like peroxides, decomposing methylglyoxal (MG), which causes fatal glycative stress, and reacting with and eliminating harmful intracellular xenobiotics [11,12]. Since selenium atoms have a higher polarizability and higher redox reactivity than sulfur atoms [13], selenoglutathione (GSeH, 1), a selenium analogue of GSH, which has Sec instead of Cys in GSH, is expected to have physiological functions far superior to those of GSH. The selenol group (-SeH) of 1 is easily oxidized by oxygen dissolved in the solvent to form a diselenide (-SeSe-) due to its high reactivity. The resulting selenoglutathione diselenide (GSeSeG, 2) is a stable compound, and the diselenide (SeSe) bond of 2 does not decompose even when left at room temperature for more than a month. This reflects the other side of the high reactivity (reducibility) of the reduced form GSeH (1) and means that selenoglutathione usually exists as the oxidized form GSeSeG (2). To bring out the unique reactivity of GSeH (1), it is necessary to activate the diselenide 2 to the selenol 1 by reduction.
Hilvert and coworkers previously reported that diselenide 2 is reduced by the enzyme glutathione reductase (GR) in the presence of NADPH to produce selenol 1 (Eq. 1) [14]. Diselenide 2 is structurally almost equivalent to GSSG, so GR recognizes 2 as a substrate. It has been reported that the substrate affinity of 2 for GR is approximately one-tenth that of GSSG [14]. On the other hand, it has recently been shown that selenol 1 can also be produced by the reaction of diselenide 2 with excess GSH (Eq. 2) [15]. These results indicate that 2 can be activated to produce 1 in cells by both enzymatic (Eq. 1) and nonenzymatic (Eq. 2) reactions.


In this review, we provide an overview of the research results on selenoglutathione reported in the literature and prospects for future research. In particular, we focus on the development of synthetic methods for selenoglutathione and the applications of the unique reactivity to biochemical reactions (Fig. 1). Section 2 describes the chemical synthesis of selenoglutathione, and Section 3 describes its formation and metabolism in vivo. Section 4 details recent research results on the applications of selenoglutathione (4.1 Protein folding, 4.2 Radical scavenging, 4.3 Detoxification of intracellular xenobiotics, 4.4 Stress reduction, and 4.5 Antibacterial activity). Section 5 briefly discusses future applications of selenoglutathione in the fields of applied biology and medicinal research based on the recent achievements.

Fig. 1.
Synthesis and applications of selenoglutathione.
2. Chemical synthesis of GSeSeG
The chemical synthesis of selenoglutathione was reported as early as the 1960s [16,17]. In the early studies, a liquid-phase method was used as the synthesis method. In 1993, Tamura [18] also used a liquid-phase method to synthesize 0.13 g of GSeSeG (2) as an ammonium salt from N-Fmoc-Se-(p-methoxybenzyl)-L-selenocysteine (Fmoc-Sec(PMB)-OH, 3) as a starting material, and succeeded in characterizing selenoglutathione by various spectra for the first time. However, the overall yield of diselenide 2 was low at 9 %, so the liquid-phase synthesis method was not used in subsequent studies for a while. In the 2000s, Beld [14] reported the chemical synthesis of 2 by a solid-phase method, which is easier to carry out experimentally (Scheme 1). They reacted activated selenocysteine (Fmoc-Sec(PMB)-OPfp) and γ-glutamic acid (Boc-Glu(OPfp)-OtBu) sequentially with Fmoc-Gly-loaded WANG resin using an automated peptide synthesizer. The tripeptide was then removed from the resin and deprotected using a trifluoroacetic acid (TFA):trimethylsilane bromide (TMSBr):thioanisole:m-cresol (750:132:120:50) cocktail to obtain diselenide 2 in an overall yield of 33 %.

Yoshida [19] used a manual solid-phase method to synthesize 2 in 9 % overall yield by sequentially reacting HOBt/DCC-activated selenocysteine (Fmoc-Sec(PMB)-OBt) and glutamic acid (Boc-Glu(OBt)-OtBu) with Fmoc-Gly-loaded Alko-PEG resin, cleaving the peptide form the resin with a TFA:H2O:phenol:thioanisole:ethanedithiol (825:50:50:50:25) cocktail, and then deprotecting the PMB group on the selenium atom of the Sec residue with TFA and 2,2'-dithiobis(5-nitropyridine) (DTNP). However, the solid-phase methods require the addition of excess amino acid reagents during amino acid coupling. In addition, the efficiency of peptide cleavage from the resin was poor, and diselenide 2 was only obtained in low yields.
Considering that selenoglutathione is a short peptide and that the synthetic raw material, a selenocysteine derivative, is expensive (see below), it seems that the liquid-phase method is more suitable for large-scale synthesis than the solid-phase method. Therefore, Shimodaira [20] reconsidered Tamura’s liquid-phase synthesis method of GSeSeG (2). As a result, they succeeded in obtaining the target product 2 from the starting material 3 in an overall yield of 90 % by the synthetic route shown in Scheme 2. First, 3 was activated with HOBt/EDCI and reacted with glycine (H-Gly-OtBu) to afford dipeptide 4 in 98 % yield. Next, the Fmoc group of 4 was deprotected with diethylamine, and then glutamic acid (Boc-Glu-OtBu), activated with HOBt/EDCI, was reacted with it to yield tripeptide 5 quantitatively. After removing the PMB group on the selenium atom with iodine, the resulting 6 was purified, and finally the other protecting groups were deprotected with 5 % H2O-TFA to afford the target compound 2. Further investigations revealed that 2 could be quantitatively obtained without going through 6 by treating tripeptide 5 with 20 % thioanisole-TFA. These synthetic methods are the most efficient of the previously reported synthetic methods for GSeSeG (2), allowing a gram sclae synthesis of 2. However, in the latter shortcut route, the deprotecting agent thioanisole may remain as an impurity even after purification and lyophilization. Therefore, the route of Scheme 2 is a steady method to obtain the target compound, although the yield is somewhat reduced.

The synthesis of selenoglutathione requires a selenocysteine derivative with appropriate protecting groups on the amino and selenol groups. The Sec derivative 3 is often used to synthesize Sec-containing peptides (selenopeptides) using the routine Fmoc method. Compound 3 is commercially available, but is expensive. It can also be synthesized from inexpensive L-serine or L-cysteine through several reaction steps and purification [21,22]. However, care must be taken as racemization may occur when selenium is introduced into the amino acid sidechain by substitution reaction [22]. It is also possible to synthesize 3 from commercially available L-selenocystine (the oxidized dimer of Sec) [23], but L-selenocystine is also expensive. In any case, an appropriate route for obtaining 3 should be selected in light of the research aims.
The synthesis of GSeH derivatives has also been reported (Fig. 2). Yonezawa [24] reported the synthesis of GSe-Me (7) and GSe-Bn (8), in which the selenium atom is protected with a Me or Bn group. Lapcinska [25] reported the synthesis of selenoglutathione derivatives 9, in which the amino group is protected with a Boc group, the carboxyl group with a tBu or Bn ester, and various coumarins are bound to the selenium atom. These derivatives are also interesting from the perspective of applying selenoglutathione in the field of biochemistry.

Fig. 2.
Synthesized derivatives of GSeH (1).
3. GSeH derivatives found in metabolites
Previously, GSeH (1) was thought not to exist in nature [26,27], but as research progressed, it became increasingly clear that 1 does exist in nature in very small amounts.
Van Dorst [28] grew Lolium perenne plants in soil containing 75SeO32– and analyzed the selenium content in the soil and plants by radiometric analysis. As a result, it was confirmed that 1 was formed in the soil. Lobinski and coworkers [29,30] suggested the presence of GSe-Me (7) and its oxidized seleninyl derivative GSeO2-Me in the metabolites of selenium-rich yeast by LC-ICP-MS analysis. Although the selenium atom is methylated, these results suggest that 1 is generated during metabolism. Rao [31] fed yeast with selenomethionine (Sem) and treated the metabolites with iodoacetic acid (IAA) or p-(hydroxymercuric)benzoic acid sodium salt (PHMB), and analyzed the products by HPLC and MS analysis. As a result, GSe-CH2CO2H and GSe-Hg(C6H4)CO2H were formed within 2 hours, indicating the production of GSeH (1). Lobinski and coworkers [32] identified 64 selenium compounds in the metabolic products of selenium-enriched yeast using reversed-phase/hydrophilic ion interaction (HILIC) liquid chromatography–electrospray hybrid quadrupole trap/Orbitrap mass spectrometry, and showed that 1 was among them. Furthermore, they estimated that the metabolic process of selenoglutathione by selenium-enriched yeast is similar to that of natural glutathione, as illustrated in Fig. 3.

Ruszczyńska [33] found GSe-Me (7) and methylthio-selenoglutathione (GSe-SMe) in garlic, sunflower sprouts and radish sprouts grown in a high-selenium medium using HPLC-ICP-MS and HPLC-ESI Orbitrap-MS/MS analysis. Similarly, the presence of GSe-SMe in the metabolites of the feed yeast Candida utilis ATCC 9950 has been reported [34]. As mentioned above, it has been revealed that selenoglutathione or its derivatives exist in the metabolites of yeast and certain plants. However, the amount of GSeH (1) is small and was observed when the organisms were grown in a medium containing a high concentration of selenium compounds. Therefore, it is more appropriate to consider it as a passive metabolic product rather than an active synthesis of GSeH (1) by the organism for particular purposes.
Recently, Gao [35] obtained a strain that synthesizes substantial amounts of GSH by mutating the yeast Saccharomyces boulardii with atmospheric and room temperature plasma (ARTP) mutagenesis, and succeeded in synthesizing a freeze-dried powder of GSeH (1), containing GSH (18 %) as an impurity, by culturing the strain in a medium containing sodium selenite (Na2SO3). The obtained powder was slightly yellow, and the structure of 1 was characterized by LC-MS analysis. This in vivo method of the synthesis is interesting as a new approach to selenoglutathione in addition to the chemical synthesis of 2 described in Section 2.
4. Applications of selenoglutathione to biochemical reactions
With the availability of GSeH (1) and GSeSeG (2), research on the reactivity of selenoglutathione and its applications in various biochemical reactions have been promoted in recent years. These pioneering studies have gradually unveiled the unique reactivity of selenoglutathione. Representative applications are described in detail below.
-
4.1. Protein folding
The first substantial application of selenoglutathione to biochemical reactions was in the oxidative folding of proteins. The research on this subject has been actively conducted by Hilvert and coworkers since the late 2000s [14,36–38]. Proteins that have disulfide bonds (SS bonds) in their molecules lose their three-dimensional structure and become denatured when the SS bonds are reduced. However, when an appropriate small molecule oxidant, such as GSSG and oxidized dithiothreitol (DTTox), is added to this denatured state (R), the protein gradually retrieves its native folded structure (N) through the formation and rearrangement of SS bonds [39]. The process from R to N is oxidative folding of proteins. The oxidant used in this process is the disulfide reagent RSSR, which has a redox potential close to that of the SS bonds of proteins. These reagents cooperatively promote the SS bond formation and SS bond rearrangement reactions at a moderate velocity, ultimately inducing the native protein (N) [40–43]. On the other hand, selenoxide reagents (>Se=O), which have strong oxidizing power, are also known to be useful for oxidative folding of proteins [44]. When selenoxide is used, the SS bond formation reaction takes precedence, followed by a slow SS bond exchange reaction, and the protein gradually folds during this latter process. A use of this reagent makes it possible to clearly determine the protein folding pathways [45].
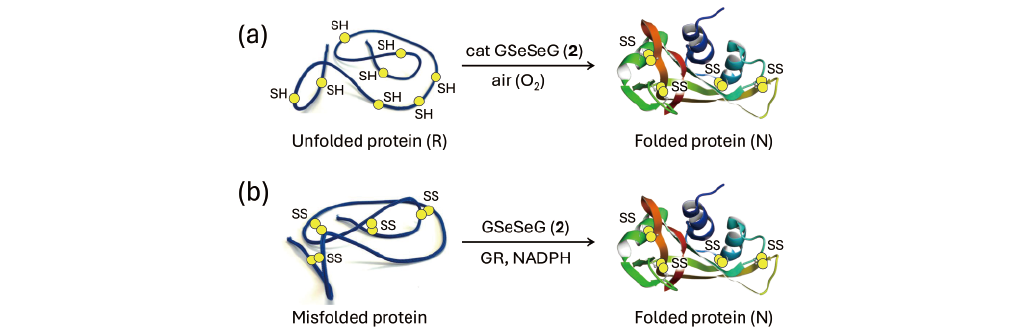
Conversely, the diselenide reagent (RSeSeR) is much less effective as an oxidizing agent than RSSR, so it is expected that RSeSeR will not promote the formation of SS bonds in proteins and will not be useful for oxidative folding. However, when GSeSeG (2) was actually applied, it was found that the folding of bovine pancreatic ribonuclease A (RNase A) and trypsin inhibitor (BPTI) proceeded faster than when GSSG was used (Fig. 4a) [14,38]. The reasons for this unexpected result would be several folds, such as because the gain in energy obtained by the formation of the three-dimensional structure of the protein exceeds the energy deficit when the R state is oxidized by GSeSeG (2). In addition, since GSeH (1) generated by the reaction between R and 2 has strong nucleophilicity, it is also assumable that 1 effectively accelerate the rearrangement of SS bonds in the folding intermediates. It was also found that GSeSeG (2) catalytically folds proteins because GSeH (1) is easily oxidized by dissolved oxygen [36]. These factors likely accelerate oxidative folding cooperatively.
GSeSeG (2) was found to be useful for the oxidative folding of various SS-containing proteins, such as hirudin, lysozyme, human epidermal growth factor, and interferon α-2a [37]. However, in the case of bovine serum albumin (BSA), which has 17 SS bonds, and antibody Fab fragments, which are considered difficult to fold, it was necessary to use high concentrations (~mM) of GSeSeG (2) [37]. In addition to 2, various small molecule diselenide reagents have been developed as useful folding reagents [46–48]. Some of them have been shown to have even better catalytic activity than 2 [46].
Unique applications that take advantage of the reaction (Eq. 1), in which GSeSeG (2) is reduced by GR to generate active GSeH (1), have also been reported. Shimodaira [20] showed in vitro that when misfolded RNase A with random SS bonds was treated with 2 in the presence of NADPH and GR, the SS bonds of the misfolded protein were reduced by the generated 1, and the rearrangement of the SS bonds proceeded to efficiently generate the N state (Fig. 4b). Thus, GSeSeG (2) would be useful for refolding of misfolded proteins.
Hilvert and coworkers [49] applied GSeSeG (2) to the in vivo oxidative folding of β-galactosidase using a DsbA-deficient E. Coli strain. As a result, the production of the native state of β-galactosidase was confirmed using 1.0 μM of 2. Thus, 2 would mediate protein folding in vivo as well as in vitro, but the effect was significantly worse than that of GSSG. Special ingenuity may be required to apply GSeSeG (2) to in vivo protein folding.
-
4.2. Radical scavenging
GSeH (1) is also useful as a radical scavenger. Radical species can cause lipid peroxidation and other oxidative stress. Koppenol and coworkers [50] used pulse radiolysis to monitor the reaction of N-acetyl-tyrosyine radical (free Tyr·) with Sec, GSeH (1), or GSH and estimated the reaction rate constants (k’s). The reaction of free Tyr· with Sec is shown in Eq. 3. The rate constant for the reaction of 1 with free Tyr· is kGSeH = 5 × 108 M-1 s-1, which is slightly smaller than that of Sec (kSec = 8 × 108 M-1 s-1), but it is 105 larger than that of GSH (kGSH = 2.4 × 103 M-1 s-1) [50]. The Sec radical generated by the reaction interacts with another Sec molecule to form a dimer radical anion with a two-center-three-electron bond (Eq. 4), which should then decompose. Interestingly, the reaction rate constant of 1 with the tyrosine radical in insulin (insulin-Tyr·) was found to be 102 smaller than that with free Tyr· [50]. This is likely due to steric hindrance caused by the folded structure of insulin. The ability of GSeH (1) as a radical scavenger is comparable to that of ascorbic acid [51].


-
4.3. Detoxification of xenobiotics
Glutathione transferases (GSTs) are enzymes that catalyze the reaction of intracellular xenobiotics (X) with GSH to form glutathione conjugates (GS-X) [52]. It has been demonstrated that GSeH (1) exerts GST-like detoxifying effects against organic mercury and allergenic aromatic chloride compounds.
Khan [53] followed the reaction of methylmercury (CH3HgOH) with 1 by real-time 1H and 199Hg NMR spectroscopy and confirmed the formation of (CH3Hg)2Se and a black precipitate of HgSe(s). Similar reactions were also observed with selenomethionine (Sem) and selenocysteine (Sec). From the analysis of the reaction products, Khan speculated that the reaction proceeded as follows (Fig. 5): Selenoglutathione (GSe–) reacts with CH3Hg+ to form GSe-HgCH3, which further reacts with GSe– to produce (CH3Hg)2Se, GSeSeG (2), and G–. (CH3Hg)2Se disproportionates to form (CH3)2Hg and HgSe(s). This study indicates that GSeH (1) may contribute to the detoxification of organic mercury.
Recently, Kanamori [15] investigated the GST-like activity of GSeSeG (2) using 1-chloro-2,4-dinitrobenzene (CDNB) as a model substance for allergenic aromatic compounds. CDNB (1 mM) and GSH (2 mM) were reacted in the presence of 2, and the amount of the resulting conjugate (GS-DNB) was determined by the change in absorbance at 340 nm (Fig. 6). The results showed that although the activity of 2 was lower than that of the GST enzyme (0.15 unit/mL), 2 exhibited significant GST-like activity in a concentration-dependent manner in the range of 0.05 to 0.5 mM.
When the reaction products were analyzed by HPLC, only one peak was observed in the presence of GST, which was identified to be GS-DNB by mass spectrometry. The same product peak was also observed in the control, indicating that the reaction between GSH and CDNB proceeded slightly even in the absence of the GST enzyme (Scheme 3). On the other hand, in the presence of 2, two peaks were observed by HPLC, and mass spectrometry confirmed that these were GS-DNB and GSe-DNB, a conjugate of selenoglutathione and CDNB. Since 2 did not react directly with CDNB, it was assumed that 2 reacted with GSH, which was present in the reaction solution, to produce 1, which was subsequently reacted with CDNB to generate GSe-DNB (Scheme 4). Thus, it was demonstrated that 1 can be generated by a nonenzymatic process between 2 and GSH (Eq. 2). This suggests that under the conditions of high GSH concentration like within cells, GSeH (1) is produced from GSeSeG (2). This is an important finding in light of biochemical applications of selenoglutathione.


Scheme 3.
Reaction of GSH with CDNB.

Scheme 4.
Reaction of GSeH (1) with CDNB.
-
4.4. Mitigation of oxidative and glycative stresses
Hydrogen peroxides (H2O2) and hypochlorous acid (HOCl) cause oxidative stress in cells. Similarly, methylglyoxal (MG) induces fatal glycative stress in cells. Recent experiments in cell-free and cell-based systems have demonstrated that selenoglutathione effectively alleviates these stresses. This section summarizes the interactions of GSeH (1) and GSeSeG(2) with H2O2, HOCl, and MG.
Glutathione peroxidase (GPx) is an antioxidant enzyme that has Sec at its active center [11,54]. This selenoenzyme uses GSH to degrade H2O2 or hydroperoxides (ROOH) generated in the body into harmless water or alcohol, respectively. Singh [55] previously reported in detail on the GPx-like antioxidant activity of Sec. In the active center of GPx, it is known that Sec does not exist alone but form a catalytic triad (or plausible tetrad) with proximate polar amino acids, such as Gln and Trp [56,57]. The interactions between these polar amino acid sidechains and the selenium atom of Sec are studied extensively, but the details of their role remain controversial [58–62].
Yoshida [19] reported that GSeSeG (2) exhibits high GPx-like antioxidant activity against H2O2 in the presence of GR and NADPH in a cell-free system. GSeH (1), which is generated from 2 by the reaction of Eq. 1, reacts rapidly with H2O2 to produce selenenic acid GSeOH and H2O. GSeOH can be further oxidaized to seleninic acid GSeO2H, but in the presence of an enough concentration of GSH, GSeOH reacts rapidly with GSH to produce selenenyl sulfide GSeSG, which is a fairly stable intermediate. As far as the reaction was traced by NMR, it could not be reduced to GSeH even when excess GSH was added [20]. However, GSeSG is structurally equivalent to GSSG, just like 2. Therefore, it would be reduced to 1 in the presence of GR and NADPH (Eq. 5). The reason why 2 exhibits high GPx-like activity is probably because the stable GSeSG intermediate is reduced by the action of GR. The updated GPx-like catalytic cycle of 2 is illustrated in Fig. 7. Shimodaira [20] used the synthesized 2 to follow a series of reactions using 77Se NMR and succeeded in characterizing some of the reaction intermediates.

Recently, Kanamori [15] attempted to quantify the remaining H2O2 in the reaction of 1 (1 mM), which was generated by the method of Eq. 1, with H2O2 (0.1 mM) by FOX assay. The FOX assay is a common method to quantify the concentration of H2O2 in a sample solution by utilizing the reaction, in which Fe2+ is oxidized to Fe3+ [63]. The reaction was performed under conditions, in which GPx (0.014 units/mL) and/or GSH (2 mM) were added to the assay solution to mimic the intracellular environment. The remaining rate of H2O2 under various reaction conditions is shown in Fig. 8.
GSeH (1) always showed a low H2O2 residual rate (<20%) regardless of the addition of GPx or GSH (2 mM). This indicates that 1 reacts directly with H2O2 through a nonenzymatic process. In contrast, the reduction of H2O2 concentration by GSH depended on the presence of both GPx and GSH (2 mM) in the assay solution. In the presence of GPx, the reduction of H2O2 by GSH is also catalyzed by the enzyme. In fact, the residual H2O2 increased in the absence of GPx. Looking at the residual H2O2 in the absence of both GPx and GSH (2 mM), it was obvious that GSH can also react directly with H2O2, but its reducing ability is significantly lower than that of GSeH (1). This reflects the fact that the selenol group (-SeH) has a stronger reducing ability than the thiol group (-SH).
Carrol [64] reported the reaction rate constants (k’s) of GSeH (1) and GSeSeG (2) with various reactive oxygen species by precise reaction kinetic analysis. The reaction rate constants of 1 and 2 with HOCl were k = 3.2 × 108 and 1.9–2.59 × 107 M-1 s-1, respectively, showing that selenoglutathione is an excellent antioxidant against HOCl. This is presumably due to the fact that selenium has a higher polarizability than sulfur.
Glyoxalase I (GLO1) is an enzyme that uses GSH to degrade methylglyoxal (MG), which causes glycative stress (Scheme 5) [65,66]. GSH reacts with MG to generate a hemithioacetal intermediate, which is then isomerized to S-lactoylglutathione by the action of GLO1. Kanamori [15] reacted GSeH (1) or GSH, which was generated from GSeSeG or GSSG, respectively, using GR and NADPH, with an equivalent amount of MG and measured the proportion of unreacted MG in the reaction solution by reacting MG with 1,2-diamino-4,5-methylenedioxybenzene dihydrochloride (MDB). The amount of the MG-MDB adduct formed was determined by measuring the fluorescence of the solution at 393 nm (Scheme 6) [67]. The results are shown in Fig. 9. It was observed that MG was rapidly consumed in the presence of 1 (approximately 50% in 30 min), whereas the reaction between GSH and MG was slow. Although this experiment did not allow the identification of the reaction product between 1 and MG, it was confirmed that 2 exhibited GLO1-like anti-glycative stress activity in the presence of GR and NADPH.
The results of the cell-free assays shown in Figs. 8 and 9 revealed that 2 can exert anti-stress activity through GPx-like and GLO1-like mechanisms in the coexistence of GR and NADPH, thus indicating that 2 is an effective enzyme mimic of GPx and GLO1 and may be applied as a precatalyst to suppress oxidative and glycative stress in cells.
It was also revealed that 2 exhibits significant antioxidative and anti-glycative stress activities in cell culture systems. Kanamori [15] pretreated HeLa cells with 2 and then MG (4 mM), exposed the cells to oxidative stress by adding H2O2 (1.2 mM), and investigated the cell viability. The results are shown in Fig. 10.
When cells were not treated with MG nor H2O2, the cell viability remained constant (about 100%) even when treated with 50 μM of 2. This indicates that 2 is non-toxic at concentrations up to at least 50 μM. This property of 2 was in significant contrast to a high toxicity of selenite (Na2SeO3) to HeLa cells (LD50 = 1.2 μM) [68]. On the other hand, when H2O2 (1.2 mM) was added to the cells, a decrease in the viability was observed for cells not pretreated with 2. This evidenced that 2 exhibits antioxidative stress activity in HeLa cells. Furthermore, when cells were pretreated with MG, the cell viability was significantly reduced due to glycative stress caused by MG. However, the cell viability was improved when the cells were pretreated with 2. Thus, 2 exhibited antioxidative and anti-glycative stress activities in the cell culture system as well as in the cell-free system, suggesting the possibility of applying 2 as an anti-stress agent.

Fig. 7.
GPx-like catalytic cycle of GSeSeG (2) updated.

Scheme 5.
Reaction between methylglyoxal (MG) and GSH in the presence of GLO1.

Scheme 6.
Reaction between methylglyoxal (MG) and MDB.
-
4.5. Antibacterial activity
As mentioned in Section 3, Gao [35] recently reported the preparation of antibacterial hydrogels using selenoglutathione. By doping GSeH (1), which was synthesized using mutant yeast, into cellulose nanocrystals together with glutathione (GSH) and biosynthesized selenium nanoparticles, an elastic hydrogel with antibacterial properties against E. coli and S. aureus was obtained. This gel also exhibited significant antioxidant and radical-scavenging capacities. The authors expected to apply the hydrogels to strain sensors. Antibacterial activities of selenoglutathione have been only little explored to date.
5. Future perspective
Selenoglutathione, a selenium analog of glutathione, was once difficult to obtain. Therefore, it was only limitedly applied to the biological research in the past. However, in recent years, chemical synthesis protocols for GSeSeG (2) using not only solid-phase [14] but also liquid-phase methods [20] have been established, and various derivatives of selenoglutathione are now available. Furthermore, GSeH (1) can be obtained by culturing mutated yeast [35]. In the future, applied research of selenoglutathione (1 and 2) thus obtained will be actively conducted. In this review, we introduced its recent applications as a protein folding agent, a radical scavenger, a detoxifier for intracellular xenobiotics, an anti-stress agent, and an antibacterial agent as the examples that have already been reported. In the application to oxidative folding of proteins, 2 is an efficient catalyst (Section 4.1). 2 is also excellent as a radical scavenger (Section 4.2) and a detoxifier for intracellular xenobiotics (Section 4.3). It has also been reported recently that 2 has stress-reducing effects (Section 4.4) and antibacterial effects (Section 4.5). Although most of these applications are basic research in cell-free systems, some have already been performed using cell systems. These pioneering studies have revealed that selenoglutathione has a much higher biological activity than the corresponding glutathione. The enhanced activity is mainly due to the fact that the selenol group of GSeH (1) is more reactive (reducible) than the thiol group.
The unique reactivity of selenoglutathione is expected to be widely applied in the fields of applied biology and medicine in the future. When applying selenium compounds to living organisms, it is necessary to evaluate their toxicity. Since GSeSeG (2) is highly water-soluble like GSSG, the toxicity of this small tripeptide dimer in vivo is expected to be low [68]. In fact, 2 showed no toxicity to HeLa cells at concentrations up to at least 50 μM [15]. The low toxicity of 2 will be a great advantage in the development of new biological functions of selenoglutathione and its application to medicines.
Acknowledgments
This research was funded by JSPS KAKENHI, grant number 22K05466 and by the Institute of Advanced Biosciences, Tokai University, research project 2024-01.
Statements about COI
There is no conflict of interest to declare.
References
- [1]
Dogaru
CB
,
Muscurel
C
,
Duță
C
,
Stoian
I
.
“Alphabet” Selenoproteins: Their Characteristics and Physiological Roles.
Int. J. Mol. Sci.
2023;
24:
15992.
- [2]
Zhang
F
,
Li
X
,
Wei
Y
.
Selenium and Selenoproteins in Health.
Biomol.
2023;
13:
799.
- [3]
Turanov
AA
,
Xu
XM
,
Carlson
BA
,
Yoo
MH
,
Gladyshev
VN
,
Hatfield
DL
.
Biosynthesis of Selenocysteine, the 21st Amino Acid in the Genetic Code, and a Novel Pathway for Cysteine Biosynthesis.
Adv. Nutr.
2011;
2:
122–128.
- [4]
Iwaoka
M
,
Shimodaira
S
.
Synthesis and catalytic functions of selenopeptides.
Organochalcogen Compounds: Synthesis, Catalysis, and New Protocols with Greener Perspectives, edited by Lenardao EJ, Santi C, Perin G, Alves D, Elsevier,
2022, Chapter 6, pp.
195-218.
- [5]
Mousa
R
,
Notis Dardashti
R
,
Metanis
N
.
Selenium and Selenocysteine in Protein Chemistry.
Angew. Chemie Int. Ed.
2017;
56:
15818–15827.
- [6]
Moroder
L
,
Musiol
HJ
.
Amino acid chalcogen analogues as tools in peptide and protein research.
J. Pept. Sci.
2020;
26:
1–15.
- [7]
Mousa
R
,
Hidmi
T
,
Pomyalov
S
,
Lansky
S
,
Khouri
L
,
Shalev
DE
,
Shoham
G
,
Metanis
N
.
Diselenide crosslinks for enhanced and simplified oxidative protein folding.
Commun. Chem.
2021;
4:30.
- [8]
Iwaoka
M
.
Chalcogen-containing Protein and Nucleic Acid Derivatives – Synthesis and Applications.
Chalcogen Chemistry: Fundamentals and Applications, edited by Lippolis V, Santi C, Lenardao EJ, Braga AL, The Royal Society of Chemistry,
2023, Chapter 24, pp.
625-647.
- [9]
Leszczynska
G
,
Cypryk
M
,
Gostynski
B
,
Sadowska
K
,
Herman
P
,
Bujacz
G
,
Lodyga-Chruscinska
E
,
Sochacka
E
,
Nawrot
B
.
C5-Substituted 2-Selenouridines Ensure Efficient Base Pairing with Guanosine; Consequences for Reading the NNG-3′ Synonymous mRNA Codons.
Int. J. Mol. Sci.
2020;
21:
2882.
- [10]
Kulik
K
,
Sadowska
K
,
Wielgus
E
,
Pacholczyk-Sienicka
B
,
Sochacka
E
,
Nawrot
B
.
Different oxidation pathways of 2-selenouracil and 2-thiouracil, natural components of transfer rna.
Int. J. Mol. Sci.
2020;
21:
1–18.
- [11]
Vašková
J
,
Kočan
L
,
Vaško
L
,
Perjési
P
.
Glutathione-Related Enzymes and Proteins: A Review.
Molecules
2023;
28:
1447.
- [12]
Georgiou-Siafis
SK
,
Tsiftsoglou
AS
.
The Key Role of GSH in Keeping the Redox Balance in Mammalian Cells: Mechanisms and Significance of GSH in Detoxification via Formation of Conjugates.
Antioxidants
2023;
12:
1953.
- [13]
Iwaoka
M
,
Arai
K
.
From Sulfur to Selenium. A New Research Arena in Chemical Biology and Biological Chemistry.
Curr. Chem. Biol.
2013;
7:
2–24.
- [14]
Beld
J
,
Woycechowsky
KJ
,
Hilvert
D
.
Selenoglutathione: Efficient oxidative protein folding by a diselenide.
Biochemistry
2007;
46:
5382–5390.
- [15]
Kanamori
A
,
Egawa
N
,
Yamasaki
S
,
Ikeda
T
,
Rocha
MJ da
,
Bortolatto
CF
,
Savegnago
L
,
Brüning
CA
,
Iwaoka
M
.
Antioxidative and Antiglycative Stress Activities of Selenoglutathione Diselenide.
Pharmaceuticals
2024;
17:
1049.
- [16]
Frank
W
.
Synthesen von selenhaltigen peptiden, II. Darstellung des Se-analogen oxydierten Glutathions (Se-Se-glutathion).
Hoppe. Seylers. Z. Physiol. Chem.
1964;
339:
214–221.
- [17]
Theodoropoulos
D
,
Schwartz
IL
,
Walter
R
.
Synthesis of Selenium-Containing Peptides.
Biochemistry
1967;
6:
3927–3932.
- [18]
Tamura
T
,
Oikawa
T
,
Ohtaka
A
,
Fujii
N
,
Esaki
N
,
Soda
K
.
Synthesis and Characterization of the Selenium Analog of Glutathione Disulfide.
Anal. Biochem.
1993;
208:
151–154.
- [19]
Yoshida
S
,
Kumakura
F
,
Komatsu
I
,
Arai
K
,
Onuma
Y
,
Hojo
H
,
Singh
BG
,
Priyadarsini
KI
,
Iwaoka
M
.
Antioxidative Glutathione Peroxidase Activity of Selenoglutathione.
Angew. Chemie Int. Ed.
2011;
50:
2125–2128.
- [20]
Shimodaira
S
,
Asano
Y
,
Arai
K
,
Iwaoka
M
.
Selenoglutathione Diselenide: Unique Redox Reactions in the GPx-Like Catalytic Cycle and Repairing of Disulfide Bonds in Scrambled Protein.
Biochemistry
2017;
56:
5644–5653.
- [21]
Iwaoka
M
,
Ooka
R
,
Nakazato
T
,
Yoshida
S
,
Oishi
S
.
Synthesis of Selenocysteine and Selenomethionine Derivatives from Sulfur-Containing Amino Acids.
Chem. Biodivers.
2008;
5:
359–374.
- [22]
Shimodaira
S
,
Iwaoka
M
.
Improved synthetic routes to the selenocysteine derivatives useful for Boc-based peptide synthesis with benzylic protection on the selenium atom.
ARKIVOC
2016;
2017:
260–271.
- [23]
Flögel
O
,
Casi
G
,
Hilvert
D
,
Seebach
D
.
Preparation of the β3-Homoselenocysteine Derivatives Fmoc-β3hSec(PMB)-OH and Boc-β3hSec(PMB)-OH for Solution and Solid-Phase-Peptide Synthesis and Selenoligation.
Helv. Chim. Acta
2007;
90:
1651–1666.
- [24]
Yonezawa
T
,
Yamaguchi
M
,
Ninomiya
M
,
Koketsu
M
.
Application of bis-2-(trimethylsilyl)ethyl diselenide to the synthesis of selenium-containing amino acid derivatives.
Tetrahedron
2017;
73:
6085–6091.
- [25]
Lapcinska
S
,
Arsenyan
P
.
Selenocysteinyl electrophiles efficiently promote the formation of coumarin and quinolinone cores by 6-endo-dig cyclization.
New J. Chem.
2021;
45:
16625–16634.
- [26]
Cowie
DB
,
Cohen
GN
.
Biosynthesis by Escherichia coli of active altered proteins containing selenium instead of sulfur.
Biochim. Biophys. Acta
1957;
26:
252–261.
- [27]
Shrift
A
,
Virupaksha
TK
.
Seleno-amino acids in selenium-accumulating plants.
Biochim. Biophys. Acta - Gen. Subj.
1965;
100:
65–75.
- [28]
Van Dorst
SH
,
Peterson
PJ
.
Selenium speciation in the soil solution and its relevance to plant uptake.
J. Sci. Food Agric.
1984;
35:
601–605.
- [29]
Dernovics
M
,
Far
J
,
Lobinski
R
.
Identification of anionic selenium species in Se-rich yeast by electrospray QTOF MS/MS and hybrid linear ion trap/orbitrap MSn.
Metallomics
2009;
1:
317–329.
- [30]
Far
J
,
Preud’homme
H
,
Lobinski
R
.
Detection and identification of hydrophilic selenium compounds in selenium-rich yeast by size exclusion-microbore normal-phase HPLC with the on-line ICP-MS and electrospray Q-TOF-MS detection.
Anal. Chim. Acta
2010;
657:
175–190.
- [31]
Rao
Y
,
McCooeye
M
,
Windust
A
,
Bramanti
E
,
D’Ulivo
A
,
Mester
Z
.
Mapping of selenium metabolic pathway in yeast by liquid chromatography-orbitrap mass spectrometry.
Anal. Chem.
2010;
82:
8121–8130.
- [32]
Arnaudguilhem
C
,
Bierla
K
,
Ouerdane
L
,
Preud’homme
H
,
Yiannikouris
A
,
Lobinski
R
.
Selenium metabolomics in yeast using complementary reversed-phase/hydrophilic ion interaction (HILIC) liquid chromatography-electrospray hybrid quadrupole trap/Orbitrap mass spectrometry.
Anal. Chim. Acta
2012;
757:
26–38.
- [33]
Ruszczyńska
A
,
Konopka
A
,
Kurek
E
,
Torres Elguera
JC
,
Bulska
E
.
Investigation of biotransformation of selenium in plants using spectrometric methods.
Spectrochim. Acta - Part B Atomic Spectrosc.
2017;
130:
7–16.
- [34]
Kieliszek
M
,
Błazejak
S
.
Speciation analysis of selenium in Candida utilis yeast cells using HPLC-ICP-MS and UHPLC-ESI-Orbitrap MS techniques.
Appl. Sci.
2018;
8:
1–14.
- [35]
Gao
F
,
Pang
Y
,
Wang
Y
,
Yang
X
,
Song
W
,
Nie
X
,
Tan
Z
,
Zhou
J
,
Xin
Y
,
Wng
D
,
Shi
H
,
Lai
C
,
Zhang
D
.
Nanocellulose/Selenoglutathione-Enhanced Antioxidant, Elastic, Antibacterial, and Conductive Hydrogels as Strain Sensors.
ACS Sustain. Chem. Eng.
2024;
12:
13622–13633.
- [36]
Beld
J
,
Woycechowsky
KJ
,
Hilvert
D
.
Catalysis of oxidative protein folding by small-molecule diselenides.
Biochemistry
2008;
47:
6985–6987.
- [37]
Beld
J
,
Woycechowsky
KJ
,
Hilvert
D
.
Diselenides as universal oxidative folding catalysts of diverse proteins.
J. Biotechnol.
2010;
150:
481–489.
- [38]
Metanis
N
,
Foletti
C
,
Beld
J
,
Hilvert
D
.
Selenoglutathione-mediated rescue of kinetically trapped intermediates in oxidative protein folding.
Isr. J. Chem.
2011;
51:
953–959.
- [39]
Arai
K
,
Iwaoka
M
.
Flexible folding: Disulfide-containing peptides and proteins choose the pathway depending on the environments.
Molecules
2021;
26:
195.
- [40]
Chatrenet
B
,
Chang
JY
.
The disulfide folding pathway of hirudin elucidated by stop/go folding experiments.
J. Biol. Chem.
1993;
268:
20988–20996.
- [41]
Wedemeyer
WJ
,
Welker
E
,
Narayan
M
,
Scheraga
HA
.
Disulfide bonds and protein folding.
Biochemistry
2000;
39:
4207–4216.
- [42]
Okumura
M
,
Saiki
M
,
Yamaguchi
H
,
Hidaka
Y
.
Acceleration of disulfide-coupled protein folding using glutathione derivatives.
FEBS J.
2011;
278:1137–1144.
- [43]
Potempa
M
,
Hafner
M
,
Frech
C
.
Mechanism of gemini disulfide detergent mediated oxidative refolding of lysozyme in a new artificial chaperone system.
Protein J.
2010;
29:
457–465.
- [44]
Arai
K
,
Dedachi
K
,
Iwaoka
M
.
Rapid and quantitative disulfide bond formation for a polypeptide chain using a cyclic selenoxide reagent in an aqueous medium.
Chem. - A Eur. J.
2011;
17:
481–485.
- [45]
Arai
K
,
Noguchi
M
,
Singh
BG
,
Priyadarsini
KI
,
Fujio
K
,
Kubo
Y
,
Tokayama
K
,
Ando
S
,
Iwaoka
M
.
A water-soluble selenoxide reagent as a useful probe for the reactivity and folding of polythiol peptides.
FEBS Open Bio
2013;
3:
55–64.
- [46]
Reddy
PS
,
Metanis
N
.
Small molecule diselenide additives for in vitro oxidative protein folding.
Chem. Commun.
2016;
52:
3336–3339.
- [47]
Arai
K
,
Ueno
H
,
Asano
Y
,
Chakrabarty
G
,
Shimodaira
S
,
Mugesh
G
,
Iwaoka
M
.
Protein Folding in the Presence of Water-Soluble Cyclic Diselenides with Novel Oxidoreductase and Isomerase Activities.
ChemBioChem
2018;
19:
207–211.
- [48]
Arai
K
,
Mikami
R
.
Redox Chemistry of Selenols and Diselenides as Potential Manipulators for Structural Maturation of Peptides and Proteins.
Metallomics Res.
2022;
2:
rev-1–rev-17.
- [49]
Beld
J
,
Woycechowsky
KJ
,
Hilvert
D
.
Small-molecule diselenides catalyze oxidative protein folding in vivo.
ACS Chem. Biol.
2010;
5:
177–182.
- [50]
Steinmann
D
,
Nauser
T
,
Beld
J
,
Tanner
M
,
Günther
D
,
Bounds
PL
,
Koppenol
WH
.
Kinetics of tyrosyl radical reduction by selenocysteine.
Biochemistry
2008;
47:
9602–9607.
- [51]
Gebicki
JM
,
Nauser
T
,
Domazou
A
,
Steinmann
D
,
Bounds
PL
,
Koppenol
WH
.
Reduction of protein radicals by GSH and ascorbate: Potential biological significance.
Amino Acids
2010;
39:
1131–1137.
- [52]
Allocati
N
,
Masulli
M
,
Di Ilio
C
,
Federici
L
.
Glutathione transferases: substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases.
Oncogenesis
2018;
7:1–15.
- [53]
Khan
MAK
,
Wang
F
.
Chemical demethylation of methylmercury by selenoamino acids.
Chem. Res. Toxicol.
2010;
23:
1202–1206.
- [54]
Pei
J
,
Pan
X
,
Wei
G
,
Hua
Y
.
Research progress of glutathione peroxidase family (GPX) in redoxidation.
Front. Pharmacol.
2023;
14:
1147414.
- [55]
Singh
BG
,
Bag
PP
,
Kumakura
F
,
Iwaoka
M
,
Priyadarsini
KI
.
Role of Substrate Reactivity in the Glutathione Peroxidase (GPx) Activity of Selenocystine.
Bull. Chem. Soc. Jpn.
2010;
83:
703–708.
- [56]
Maiorino
M
,
Aumann
KD
,
Brigelius-Flohe
R
,
Ursini
F
,
van den Heuvel
J
,
McCarthy
J
,
Roveri
A
,
Ursini
F
,
Flohé
L
.
Probing the Presumed Catalytic Triad of Selenium-Containing Peroxidases by Mutational Analysis of Phospholipid Hydroperoxide Glutathione Peroxidase (PHGPx).
Biol. Chem. Hoppe. Seyler.
1995;
376:
651–660.
- [57]
Tosatto
SCE
,
Bosello
V
,
Fogolari
F
,
Mauri
P
,
Roveri
A
,
Toppo
S
,
Flohé
L
,
Ursini
F
,
Maiorino
M
.
The catalytic site of glutathione peroxidases.
Antioxidants Redox Signal.
2008;
10:
1515–1525.
- [58]
Iwaoka
M
,
Tomoda
S
.
A Model Study on the Effect of an Amino Group on the Antioxidant Activity of Glutathione Peroxidase.
J. Am. Chem. Soc.
1994;
116:
2557–25561.
- [59]
Sarma
BK
,
Mugesh
G
.
Glutathione peroxidase (GPx)-like antioxidant activity of the organoselenium drug ebselen: Unexpected complications with thiol exchange reactions.
J. Am. Chem. Soc.
2005;
127:11477–11485.
- [60]
Orian
L
,
Mauri
P
,
Roveri
A
,
Toppo
S
,
Benazzi
L
,
Bosello-Travain
V
,
Palma
AD
,
Maiorino
M
,
Miotto
G
,
Zaccarin
M
,
Polimeno
A
,
Flohé
L
,
Ursini
F
.
Selenocysteine oxidation in glutathione peroxidase catalysis: an MS-supported quantum mechanics study.
Free Radic. Biol. Med.
2015;
87:
1–14.
- [61]
Masuda
R
,
Kimura
R
,
Karasaki
T
,
Sase
S
,
Goto
K
.
Modeling the Catalytic Cycle of Glutathione Peroxidase by Nuclear Magnetic Resonance Spectroscopic Analysis of Selenocysteine Selenenic Acids.
J. Am. Chem. Soc.
2021;
143:
6345–6350.
- [62]
Iwaoka
M
,
Oba
H
,
Matsumura
K
,
Yamanaka
S
,
Shimodaira
S
,
Kusano
S
,
Asami
T
.
Antioxidant Activity of a Selenopeptide Modelling the Thioredoxin Reductase Active Site is Enhanced by NH···Se Hydrogen Bond in the Mixed Selenosulfide Intermediate.
Curr. Chem. Biol.
2022;
16:
44–53.
- [63]
Watanabe
N
,
Forman
HJ
.
Autoxidation of extracellular hydroquinones is a causative event for the cytotoxicity of menadione and DMNQ in A549-S cells.
Arch. Biochem. Biophys.
2003;
411:
145–157.
- [64]
Carroll
L
,
Gardiner
K
,
Ignasiak
M
,
Holmehave
J
,
Shimodaira
S
,
Breitenbach
T
,
Iwaoka
M
,
Ogilby
PR
,
Pattison
DI
,
Davies
MJ
.
Interaction kinetics of selenium-containing compounds with oxidants.
Free Radic. Biol. Med.
2020;
155:
58–68.
- [65]
Thornalley
PJ
.
Glyoxalase I – structure, function and a critical role in the enzymatic defence against glycation.
Biochem. Soc. Trans.
2003;
31:
1343–1348.
- [66]
Distler
MG
,
Palmer
AA
.
Role of glyoxalase 1 (Glo1) and methylglyoxal (MG) in behavior: Recent advances and mechanistic insights.
Front. Genet.
2012;
3:
250.
- [67]
Ogasawara
Y
,
Tanaka
R
,
Koike
S
,
Horiuchi
Y
,
Miyashita
M
,
Arai
M
.
Determination of methylglyoxal in human blood plasma using fluorescence high performance liquid chromatography after derivatization with 1,2-diamino-4,5-methylenedioxybenzene.
J. Chromatogr. B
2016;
1029–1030:
102–105.
- [68]
Richard
MJ
,
Guiraud
P
,
Didier
C
,
Seve
M
,
Flores
SC
,
Favier
A
.
Human Immunodeficiency Virus Type 1 Tat Protein Impairs Selenoglutathione Peroxidase Expression and Activity by a Mechanism Independent of Cellular Selenium Uptake: Consequences on Cellular Resistance to UV-A Radiation.
Arch. Biochem. Biophys.
2001;
386:
213–220.

 https://ror.org/00hhkn466
https://ror.org/00hhkn466 


