Abstract
This review highlights the unique chemistry of π-bond compounds consisting of both selenium and other heavier main-group element, which exhibit intriguing reactivity and potential applications in catalysis, materials science, and bioinorganic chemistry. While selenium shares similarities with sulfur, its larger atomic size and greater polarizability give Se-containing double bonds distinctive characteristics that often result in higher reactivity. This review will focus on recent advancements of compounds that contain a π-bond containing selenium and other heavier main-group elements as well as unique π-conjugated systems, rather than on well-established compounds such selenoketones and selenoamides.
1. Introduction
-
1.1 General introduction
Selenium, a member of the chalcogen family (group-16 elements), exhibits a rich and versatile chemistry that has garnered significant attention in recent decades[1]. Among its various bonding modes, the formation of double bonds involving selenium is of particular interest due to its implications in a wide array of chemical and biological processes. Indeed, compounds featuring selenium double bonds offer intriguing properties, including unique reactivity patterns and potential applications in catalysis, materials science, and bioinorganic chemistry[1,2]. Despite the structural similarities between selenium and sulfur, the chemistry of selenium double bonds reveals distinct characteristics arising from the larger atomic size, lower electronegativity, and greater polarizability of selenium. These factors influence the stability, electronic properties, and reactivity of selenium-containing double-bond systems. As a result, selenium double bonds often exhibit reactivity higher than their sulfur counterparts, unlocking avenues for the development of new synthetic methodologies.
This review aims to provide a comprehensive overview of recent advancements in the study of selenium double-bond compounds, focusing on their synthesis, structural features, and fundamental reactivity. By comparing the behavior of selenium-based double-bond systems with systems that contain other group-16 elements, we seek to highlight the unique features of selenium chemistry and its potential for further exploration in both fundamental and applied research. It should be noted here that simple double-bond systems that contain selenium and second-row-elements such as selenoketones and selenoamides have already been introduced and discussed in several reviews/books[2,3], and will thus not be considered in this review. This mini-review focuses on recent examples of compounds with double bonds that contain selenium and other heavier main-group elements as well as unique π-conjugated systems that include selenium. These reactive Se-containing double-bond systems are often stabilized by intermolecular coordination from an electron-donor, which electronically perturbs the intrinsic nature of the Se-π bonds.
-
1.2. Theoretical aspects
For many years, it had been commonly accepted that compounds that contain double bonds between heavier main-group elements are most likely unstable species due to their substantially higher reactivity compared to that of their second-row-element analogues (“double-bond rule”)[4]. Although several examples of C=S and C=Se double-bond compounds had been isolated and characterized, examples of selenium-containing double-bond compounds with a heavier main-group element remained elusive. However, following the pioneering reports on the synthesis and isolation of the first stable disilene[5] and diphosphene[6] in 1981 by the groups of West and Yoshifuji, respectively, it was demonstrated that compounds with double bonds between heavier main-group elements can be isolated as stable compounds when sufficiently stabilized by sterically demanding substituents (kinetic stabilization)[7]. After this breakthrough, the so-called heavy ketones (E=Ch double-bond compounds; E = Si, Ge, Sn, Ch = S, Se, Te), i.e., the heavier-element analogues of ketones, were successfully synthesized, isolated, and fully characterized[8].
The predominant reason for the difficulties associated with the isolation of compounds that contain a double bond between heavier main-group elements is that the bonding energy of a π-bond between heavier main-group elements is very small relative to the corresponding σ-bond energy, which causes facile oligomerization even under inert atmospheres. As shown in Table 1-1[9], the energy of the Se=E (E = C, Si, Ge, Sn, Pb) π-bonds is significantly lower than the energy of the corresponding σ-bonds and of the (S=E) π-bond energy of the corresponding sulfur analogues.
Natural-bond-orbital (NBO) calculations on the bonding character of Se=E double bonds (E = group 13-15 elements)[10], suggested that the Se=E σ-bonds should be composed of a –60% contribution from the Se atom with its spn hybridized orbital (n > 4.7), reflecting the intrinsic nature of unfavorable formation of hybridized orbitals in heavier main-group elements. The π-bonds of Se=E should consist predominantly of contributions from the p-orbital of the Se atom (>60%) probably due to the larger effective nuclear charge for the valence orbitals of the Se atom. Accordingly, the Se atoms should be negatively charged relative to the bonding E atoms. It should also be noted here that a nucleophile, such as a phosphine or alkyl lithium, sometimes attacks the Se atom of Se=E compounds despite the negative charge of the Se atom, which is probably due to the low-lying π* orbitals of the Se=E bonds that spreading largely around the Se atom. This bonding behavior should result in amphiphilic character of the Se atoms in Se=E compounds.
Given that there have recently been reported several examples of Se-containing π-bond compounds with heavier main-group elements, which have been isolated by using kinetic and/or thermodynamic stabilization, we will introduce in the next section the synthesis and structural features of these examples.
Table 1-1.
Energy values (kcal/mol) and lengths (Å) of the σ-bond energy and π-bond energy of Ch=EH
2 (E = C, Si, Ge, Sn, Pb) compounds.
| Ch=EH2 |
|
Ch = O |
Ch = S |
Ch = Se |
Ch = Te |
| E = C |
σ-BEa
|
93.6 |
73.0 |
65.1 |
57.5 |
| π-BEb
|
95.3 |
54.6 |
43.2 |
32.0 |
| distancec
|
1.200 |
1.617 |
1.758 |
1.949 |
| E = Si |
σ-BEa
|
119.7 |
81.6 |
73.7 |
63.2 |
| π-BEb
|
58.5 |
47.0 |
40.7 |
32.9 |
| distancec
|
1.514 |
1.945 |
2.082 |
2.288 |
| E = Ge |
σ-BEa
|
101.5 |
74.1 |
67.8 |
59.1 |
| π-BEb
|
45.9 |
41.1 |
36.3 |
30.3 |
| distancec
|
1.634 |
2.042 |
2.174 |
2.373 |
| E = Sn |
σ-BEa
|
94.8 |
69.3 |
64.3 |
56.4 |
| π-BEb
|
32.8 |
33.5 |
30.6 |
26.3 |
| distancec
|
1.802 |
2.222 |
2.346 |
2.543 |
| E = Pb |
σ-BEa
|
80.9 |
60.9 |
57.0 |
50.3 |
| π-BEb
|
29.0 |
30.0 |
27.8 |
24.4 |
| distancec
|
1.853 |
2.273 |
2.394 |
2.590 |
a Ch=E s-bond energy (kcal/mol); b Ch=E p-bond energy (kcal/mol); c Ch=E distance (Å).
Table 1-2.
Bonding characters, bond lengths (Å), and natural charges for Ch=EH
2 (E = C, Si, Ge, Sn, Pb) compounds calculated at the B3PW91-D3(BJ)/def2TZVPP level using Gaussian 16 (rev. C) and NBO 7.0.
E=Se (Å)
σ: E + Se
π: E + Se
NPA: E, Se
|
group-13
HE=Se (E = B, Al, Ga, In, Tl)
|
group-14
H2E=Se (E = C, Si, Ge, Sn, Pb)
|
| 2nd row
|
E = B: HB=Se: 1.7279
σ: B(sp1.04, 43.5%)+Se(sp2.88, 56.5%)
π: B(p, 25.3%)+Se(p, 74.7%)
π: B(p, 25.3%)+Se(p, 74.7%)
NPA: B(+0.15), Se(–0.10)
|
E = C: H2C=Se: 1.7441
σ: C(sp1.80, 58.1%)+Se(sp4.67, 41.8%)
π: C(p, 46.3%)+Se(p, 53.7%)
NPA: C(–0.54), Se(+0.16)
|
| 3rd row
|
E = Al: HAl=Se: 2.1238
σ: Al(sp0.89, 31.5%)+Se(sp4.88, 68.5%)
π: Al(p, 14.0%)+Se(p, 86.0%)
π: Al(p, 14.0%)+Se(p, 86.0%)
NPA: Al(+1.13), Se(–0.78)
|
E = Si: H2Si=Se: 2.0743
σ: Si(sp1.67, 41.2%)+Se(sp4.72, 58.8%)
π: Si(p, 28.3%)+Se(p, 71.7%)
NPA: Si(+0.79), Se(–0.45)
|
| 4th row
|
E = Ga: HGa=Se: 2.1252
σ: Ga(sp0.91, 36.1%)+Se(sp5.19, 63.8%)
π: Ga(p, 14.3%)+Se(p, 85.7%)
π: Ga(p, 14.3%)+Se(p, 85.7%)
NPA: Ga(+0.93), Se(–0.69)
|
E = Ge: H2Ge=Se: 2.1529
σ: Ge(sp1.69, 43.0%)+Se(sp5.84, 57.0%)
π: Ge(p, 28.1%)+Se(p, 71.9%)
NPA: Ge(+0.74), Se(–0.45)
|
| 5th row
|
E = In: HIn=Se: 2.2979
σ: In(sp0.85, 36.9%)+Se(sp8.20, 63.2%)
π: In(p, 12.1%)+Se(p, 87.9%)
π: In(p, 12.1%)+Se(p, 87.9%)
NPA: In(+1.03), Se(–0.74)
|
E = Sn: H2Sn=Se: 2.3290
σ: Sn(sp1.57, 42.3%)+Se(sp7.70, 57.7%)
π: Sn(p, 23.7%)+Se(p, 76.3%)
NPA: Sn(+0.95), Se(–0.57)
|
| 6th row
|
E = Tl: HTl=Se: 2.3322
σ: Se(sp10.5)→Tl(p) (coordination)
π: Tl(p, 11.4%)+Se(p, 88.6%)
π: Tl(p, 11.4%)+Se(p, 88.6%)
NPA: Tl(+0.90), Se(–0.68)
|
E = Pb: H2Pb=Se: 2.4040
σ: Pb(sp1.47, 46.9%)+Se(sp11.28, 53.1%)
π: Pb(p, 21.2%)+Se(p, 78.8%)
NPA: Pb(+0.87), Se(–0.54)
|
E=Se (Å)
σ: E + Se
π: E + Se
NPA: E, Se
|
group-15
HE=Se (E = N, P, As, Sb, Bi)
|
group-16
E=Se (E = O, S, Se, Te, Po)
|
| 2nd row
|
E = N: HN=Se: 1.7110
σ: N(sp3.42, 64.1%)+Se(sp5.96, 35.9%)
π: N(p, 56.2%)+Se(p, 43.8%)
NPA: N(–0.74), Se(+0.42)
|
E = O: O=Se: 1.6401
σ: O(sp4.41, 69.3%)+Se(sp6.81, 30.7%)
π: O(p, 65.7%)+Se(p, 34.3%)
NPA: O(–0.68), Se(+0.68)
|
| 3rd row
|
E = P: HP=Se: 2.0718
σ: P(sp5.37, 45.8%)+Se(sp5.95, 54.2%)
π: P(p, 41.5%)+Se(p, 58.5%)
NPA: P(+0.25), Se(–0.18)
|
E = S: S=Se: 2.0380
σ: S(sp6.85, 53.1%)+Se(sp7.71, 46.9%)
π: S(p, 53.2%)+Se(p, 46.8%)
NPA: S(–0.12), Se(+0.12)
|
| 4th row
|
E = As: HAs=Se: 2.1823
σ: As(sp7.07, 44.0%)+Se(sp6.71, 56.0%)
π: As(p, 39.9%)+Se(p, 60.1%)
NPA: As(+0.35), Se(–0.26)
|
E = Se: Se=Se: 2.1737
σ: Se(sp8.88, 50.0%)+Se(sp8.88, 50.0%)
π: Se(p, 50.0%)+Se(p, 50.0%)
NPA: Se(0.0), Se(0.0)
|
| 5th row
|
E = Sb: HSb=Se: 2.3594
σ: Sb(sp7.98, 40.1%)+Se(sp7.64, 59.9%)
π: Sb(p, 35.2%)+Se(p, 64.8%)
NPA: Sb(+0.61), Se(–0.43)
|
E = Te: Te=Se: 2.3628
σ: Te(sp10.20, 45.4%)+Se(sp9.95, 54.6%)
π: Te(p, 44.8%)+Se(p, 55.2%)
NPA: Te(+0.19), Se(–0.19)
|
| 6th row
|
E = Bi: HBi=Se: 2.4427
σ: Bi(sp11.7, 39.4%)+Se(sp8.81, 60.6%)
π: Bi(p, 34.2%)+Se(p, 65.8%)
NPA: Bi(+0.66), Se(–0.47)
|
E = Po: Po=Se: 2.4529
σ: Po(sp14.81, 44.2%)+Se(sp11.24, 55.8%)
π: Po(p, 43.4%)+Se(p, 56.6%)
NPA: Po(+0.25), Se(–0.25)
|
2. Group-13 – Selenium π-bond compounds

Figure 2-1.
Isolobal relationship between the carbonyl group and its heavier main-group analogues.

Figure 2-2.
Pioneering studies on a) oxoborane A, b) thioxoborane B, and c) oxoborane complex C.
Due to the synthetic importance of the carbonyl group, the synthesis of their main-group analogues containing a multiple bond to a group-16 element (Ch = O, S, Se, Te) has been investigated extensively. As group-13 elements (E13 = B, Al, Ga, In, Th) are more electropositive than carbon, the resulting E13=Ch bonds are highly polarized compared to the carbonyl C=O bond (Figure 2-1). Unfortunately, the isolation of compounds with E13=Ch bonds is usually difficult due to self-oligomerization. A pioneering study on transient oxoborane A was reported by West and co-workers, who generated it via the irradiation of 1,3-dioxa-2,4-boretane[11]. The elusive nature of A indicates that the bulky aryl group, Mes*, is insufficient to kinetically stabilize this highly reactive oxoborane species at ambient temperature (Figure 2-2a). The first example of compounds with an E13=S bond, i.e., thioxoborane B with a bulky aryl substituent, was reported by Okazaki and co-workers in 1996 (Figure 2-2b)[12]. In 2005, Cowley and co-workers reported that the isolation of the first example of a monomeric stable oxoborane bearing a B=O moiety (C)[13]. The compound was synthesized using a β-diketiminate as a supporting ligand for boron, and it was stabilized by the addition of the Lewis acid AlCl3. Further investigations led to the synthesis of several compounds that contain E13=Ch multiple bonds[14,15]. However, examples of E13=Se compounds are more scarce compared to other chalcogenides due to the high propensity for dimerization of the E13=Se bond. In this chapter, the synthesis and properties of compounds with an E13=Se bond are described.
-
2.1. B=Se compounds
In 2010, Cui and co-workers reported the synthesis of stable selenoxoborane 1 as orange crystals from the reaction of hydroborane 2, which bears a β-diketiminate ligand, with 1 equivalent of elemental selenium at 70 °C in toluene[16]. The formation mechanism was interpreted in terms of a B-H bond insertion of selenium, followed by an H-migration to the exocyclic methylene group (Figure 2-3a). Selenoxoborane 1 is extremely air- and moisture-sensitive. The 11B NMR spectrum of 1 exhibits a broad resonance at 40.88 ppm, which is downfield-shifted relative to that of 2 (29.3 ppm). The 77Se NMR resonance for 1 is observed at -196 ppm. The molecular structure of 1 in the crystalline state revealed that the length of the B-Se bond (1.896(4) Å) is comparable to Pyykkö’s standard value for a B-Se double bond (1.85 Å), and significantly shorter than those of reported B-Se single bonds (1.960-2.13 Å) (Figure 3b)[17-22]. The central boron atom adopts an approximately trigonal-planar coordination geometry. These structural features are consistent with a B-Se multiple bond. The bonding situation was furthermore analyzed by theoretical calculations on the Kohn-Sham orbitals of 1, which revealed that the HOMO corresponds predominantly to the selenium lone pair and that the HOMO-1 refers to the B-Se π-bond.
Singh and co-workers have obtained bis(phosphinimino)amide-substituted selenoxoborane 3 from the reaction of a hydroborane with elemental selenium (Figure 2-4a)[23]. Compound 3 exhibits structural features and properties similar to those of 1. The 11B NMR spectrum of 3 shows a broad singlet resonance at 45.2 ppm, similar to the shift for 1 (40.88). The length of the B-Se bond (1.871(5) Å) is almost identical to that of 1 (1.896(4) Å).
Trzaskowski and Frank have reported the synthesis of a series of chalcogenoxoboranes (4-7) using the same NacNac-type ligand, i.e., HAmIm (Figure 2-5)[24]. Oxoborane 4 was obtained from the reaction of bromoborane 8 with H2O to give the hydroxyborane 9, followed by a deprotonation reaction with KHMDS in the presence of [2.2.2]cryptand. Heavier B=Ch compounds 5-7 were synthesized by the reaction of bromoborane 8 with the corresponding lithium chalcogenides (Li2Ch; Ch = S, Se, Te). The 11B NMR spectra of 4-7 show similar values (21.4 ppm for 4, 35.2 ppm for 5, 35.8 ppm for 6, and 30.2 ppm for 7). The crystal structures of 4-7 revealed that all compounds contain a boron center with a trigonal-planar coordination geometry and B-Ch double-bond character. The length of the B=Se bond in 7 (1.909(2) Å) is the longest B=Se distance reported so far. The Wiberg bond indices (WBIs) decrease continuously with increasing atomic number of the chalcogen element from 1.86 in 4 to 1.69 in 7, which indicates diminished π-π interactions between the boron atom and the heavier chalcogen atoms. Meanwhile, the values for 4-7 (>1.60) justify the formulation of the B=Ch bonds as true double bonds.
Braunschweig and co-workers have reported the synthesis of N-heterocycliccarbene (NHC)-stabilized selenoxoborane 10 via the deselenization of boradiselenirane 11, which was obtained from the reaction of borylene complex 12 with elemental selenium (Figure 2-6)[25]. At 60 °C, 10 slowly dimerizes (48 h) to give four-membered cycle 13. The 11B NMR spectrum of 10 exhibits a broad resonance at -2.9 ppm, which is upfield-shifted relative to that of 1 (40.88 ppm). The 77Se NMR resonance of 10 is observed at 250 ppm, which is significantly downfield-shifted compared to that of 1 (-196 ppm) and upfield-shifted relative to that of previous reported selenoxoborane Mn complex 14 (370 ppm)[26]. The crystal structure of 10 revealed a length of the B-Se bond (1.876(4) Å) and a coordination geometry (trigonal planar) of the boron atom that are typical for selenoxoboranes.

Figure 2-3.
a) Synthesis of stable selenoxoborane 1. b) Molecular structure of 1 in the crystalline state. c) Representations of the frontier orbitals of 1.

Figure 2-4.
Synthesis of stable selenoxoborane 3. b) Molecular structure of 3 in the crystalline state.

Figure 2-5.
a) Synthesis and b) crystal structures of chalcogenoboranes 4-7.

Figure 2-6.
a) Synthesis of selenoxoborane 10, its dimer 13, and Mn complex 14. b) Crystal structure of 13.
-
2.2. Al=Se compounds
In 2023, Braunschweig and co-workers reported the synthesis of neutral aluminum selenide 16 by reacting NHC-stabilized alumylene 15 with elemental selenium in almost quantitative yield (Figure 2-7)[27]. Inoue and co-workers had previously attempted the synthesis of a neutral aluminum selenide, but obtained only the corresponding dimer[28]. The 77Se NMR spectrum of 16 exhibits a characteristic resonance signal at -264 ppm, which is up-field shifted compared to those of the compounds with B=Se bonds (Table 1). The crystal structure of 16 revealed that the central Al atom adopts a tricoordinate distorted trigonal-planar coordination geometry with an angle sum around Al of 359.8°. The Al-Se bond (2.1935(8) Å) is currently the shortest reported Al-Se bond, and comparable to Pyykkö’s standard value for an Al-Se double bond (2.20 Å). The WBI for the Al-Se bond in 16 (1.31) is smaller than that for tellurium derivative 17 (1.47). The Kohn-Sham orbitals of 16 show that the HOMO corresponds mainly to the selenium lone pair and the HOMO-1 to the Al-Se π-bond (Figure 2-7c). The NBO charges of Al (+1.56 for 16, +1.40 for 17) and Ch (–1.05 for 16, –0.89 for 17) suggest a considerable amount of charge separation. The NBO results suggest that the Al-Se bond is more polar than the Al-Te bond, which is consistent with the fact that the electronegativity of Se (2.55) is higher than that of Te (2.12). Furthermore, an intrinsic-bond-orbital (IBO) analysis indicated the presence of a single Al-Ch σ-bond, strengthened by the electrostatic attraction between the Al+ and Se– centers as well as a slight donation from the lone pairs of Se to the vacant orbitals at Al. These results suggest multiple-bond character for the Al-Se bond, albeit with an ambiphilic nature.
The reactivity of 16 corroborates its proposed ambiphilic multiple-bond character (Figure 2-8). The reaction of 16 with adamantylazide (AdN3) in toluene at room temperature rapidly afforded 18 with a three-membered Al-N-Se cycle, which is the first example of an aluminum and selenium containing analog of aziridine (Figure 2-8a). Compound 18 is likely formed via a [3 + 2] cycloaddition of AdN3 with 16 followed by N2 elimination. Furthermore, 16 can activate small molecules such as methyl iodide (MeI), phenylsilane (PhSiH3), and perfluorobenzene (C6F6), whereby the corresponding C–I, Si–H, and C–F bonds are cleaved to furnish addition products 19, 20, and 21, respectively.
In 2019, Coles and co-workers reported the synthesis of anionic selenoxoalumane [K(THF)][Al(NONAr)(Se)] (NONAr = [O(SiMe2NAr)2]2-, Ar = 2,6-i-Pr2C6H3) (23) by reacting aluminyl anion Al(NONAr)– (22) with elemental selenium (Figure 2-9a)[29]. The crystal structure of 23 revealed the formation of an infinite chain structure by linking K+ cations through Se···K and π-arene interactions. In the crystal, the aluminum center adopts a three-coordinate distorted trigonal-planar coordination environment with a short Al-Se bond (2.225(1) Å). The selenium atom interacts with two solvated potassium cations (Se···K: 3.2965(10) Å/3.3730(10) Å) to form a one-dimensional chain. To isolate the aluminum-selenium bond from Se···K interactions, the reaction was performed in the presence of [2.2.2]cryptand, which provided selenoxoalumane 24, wherein the potassium cation is fully solvated by the [2.2.2]cryptand. The Al–Se bond in 24 (2.2032(6) Å) is shorter than that of 23, confirming that the Se···K interactions in 24 cause a slight elongation of the bond. The WBI for the Al-Se bond in 24 (1.39) is slightly higher than that of 16 (1.31), which indicates the presence of significant multiple-bond character in 24. The NBO charges in 24 for Al (+1.61) and Se (–1.06) are indicative of a slightly higher polarization of the Al–Se bond in 24 compared to that in 16. The spectroscopic analysis of 23 was limited by its low solubility and/or instability in common solvents, whereas 24 allowed for a successful analysis due to its improved solubility. The 77Se NMR spectrum of 24 displays a singlet at -563 ppm, which is upfield-shifted compared to those of reported E13=Se compounds.
Treatment of selenoxoalumane 24 with an equimolar amount of elemental selenium in THF afforded diselenirane 25 as bright purple crystals (Figure 2-10).

Figure 2-7.
Synthesis of stable selenoxoalumane 16 and telluroxoalumane 17. b) Crystal structure of 16. c) Kohn-Sham frontier orbitals of 16.

Figure 2-8.
Reactivity of selenoxoborane 16.

Figure 2-9.
a) Synthesis and b,c) crystal structures of anionic selenoxoalumanes 23 and 24.

Figure 2-10.
Reactivity of selenoxoalumane 24.
3. Group-14 – Selenium π-bond compounds
Arguably the most common molecules containing a π-bond between group-14 elements (E14 = C, Si, Ge, Sn, Pb) and group-16 elements (Ch = O, S, Se, Te) are ketones. Heavy ketones (R2E14=Ch), which feature a double bond between heavier group-14 and -16 elements, are attractive synthetic targets due to their unique properties and high reactivity. Among these, selenium analogues are particularly valuable because their electronic state in solution can be analyzed using 77Se NMR spectroscopy. Unfortunately, their isolation is usually difficult due to self-oligomerization (as in the case of E13=Ch species; vide supra). The study of selenium-containing heavy ketones has a long history, beginning with Burton and co-workers, who reported in 1975 the synthesis of di-t-butylselenoketone (D) as the first monomeric compound of this class (Figure 3-1)[30]. Subsequently, in 1989, Corriu and co-workers reported the first silaneselone derivatives (E) stabilized by an intramolecular N→Si donor-acceptor interaction (thermodynamic stabilization)[31]. This method, remains widely used today and has become one of the strongest tools for stabilizing highly reactive species of main-group elements. Another primary method for stabilizing reactive main-group elements species is the previously mentioned kinetic stabilization by taking advantage of steric protection groups. Pioneering work on stable heavy ketones with a terminal group-16 element has been reported mainly by Okazaki and Tokitoh through the introduction of bulky 2,4,6-tris(bis(trimethylsilyl)methyl)phenyl and 2,6-bis(bis(trimethylsilyl)methyl)-4-(tris(trimethylsilyl)methyl)phenyl groups to group-14 elements[8]. The Ge-Se bond in germaneselone F (2.180(2) Å) is comparable to Pyykkö’s standard value for a Ge-Se double bond (2.18 Å)[17,32], while, those of thermodynamically stabilized germaneselones show slightly longer (2.194(1) Å-2.237(2) Å) due to the effect of zwitterionic canonical structures (Figure 3-1b)[33-54]. However, such base-stabilization (thermodynamic stabilization) provides access only to a relatively limited range of heavier analogues of common, small organic molecules such as ketones. In this chapter, notable achievements of new types of E14=Se species, such as heavier analogues of carboxylic acids, acylium ions, carbon dioxide, and carbonyl ylides, are described.

Figure 3-1.
Pioneering studies on a) selenoketone D, b) silaneselone E, and c) germaneselone F.
-
3.1. Heavier analogues of carboxylic acids
In 2004, Roesky and co-workers reported the synthesis of thiogermanoic acid 28 as the first heavier carboxylic acid, by reacting hydroxygermylene 27 with elemental sulfur (Figure 3-2)[55]. In 2006, selenogermanoic acid 29 was obtained by the same manner using elemental selenium instead of elemental sulfur[37]. The OH stretching frequency of 29 shows a broad absorption at 3299 cm-1, which is at slightly higher wavenumber than that of 28 (3238 cm-1). These values are significantly shifted to lower wavenumbers compared to that of 27 (3571 cm-1), indicating the formation of hydrogen bonds in 28 and 29. In the 1H NMR spectra of 28 and 29, their OH protons were observed at 2.30 ppm and 2.19 ppm, respectively, which are significantly upfield-shifted compared to carboxylic acids, indicating the weaker acidity for 28 and 29 than for common carboxylic acids. The 77Se NMR resonance of 29 is observed at –439 ppm, which falls within the range of compounds exhibiting ylide-type and multiple-bond character at the germanium-selenium moiety[56]. In the crystal, 29 exists as a selenoxo tautomeric form of hydrogen-bonded dimers with weak intermolecular hydrogen interactions. The length of the Ge-Se bond in 29 (2.206(1) Å) is comparable to Pyykkö’s standard value for a Ge-Se double bond (2.18 Å), and very much consistent with resonance-structure contributions from both a Ge-Se ylide-type bond and a multiple bond rather than a pure germanium-selenium single bond (2.37 Å)[17]. The pKa values of 28 (37.2) and 29 (38.3) fall in the range expected for aromatic (–33) and aliphatic (–48) compounds. Importantly, these values are significantly lower than those observed for representative oxygen-containing Brønsted acids (–15) [57].
Subsequently, in 2013, Driess and co-workers reported the synthesis of the stable selenosilanoic acid-base adduct of the type [LSi(Se)=OH(dmap)] (32) by reacting silanone complex 30 with H2Se (Figure 3-3)[58]. Compound 32 is the heavier analogue of a carboxylic acid and its sulfur analogue of the type [LSi(S)=OH(dmap)] (31), which had been reported by the same group in 2010[59]. In the 1H NMR spectra of 31 and 32 their OH protons are observed at 6.35 and 6.42 ppm, respectively, which are significantly downfield-shifted compared to those of germanium derivatives 28 and 29. The 77Se NMR resonance of 32 is observed at –545 ppm, which is similar to that of germanium derivative 29. The crystal structures of 31 and 32 show that they are acid-base complexes, wherein one dmap ligand is connected to the chalcogenosilanoic-acid moiety through an O-H···N hydrogen bond. The length of the Si-Se bond in 32 (2.1348(7) Å) is close to Pyykkö’s standard value for a Si-Se double bond (2.14 Å)[17].

Figure 3-2.
a) Synthesis of chalcogenogermanoic acids 28 and 29 as well as packing structures for b) 28 and c) 29.

Figure 3-3.
a) Synthesis of chalcogenosilanoic acids 31 and 32 and b) crystal structure of selenosilanoic acid 32.
-
3.2. Heavier analogues of acylium ions
Koley and Inoue have reported the synthesis of NHC-stabilized germa-acylium ion 34 by reacting germyliumylidene ion 33 with N2O[60]. Subsequently, the reaction of 34 with Lawesson’s reagent (LR) and Woollins’ reagent (WR) afforded the corresponding sulfur (35) and selenium analogues (36). The driving force of these reactions is the formation of a stable P=O bond in a cycloreversion step that resembles the mechanism of Wittig reactions, indicating that the reactivity of germa-acylium ion 34 is akin to classical acylium-like behavior. The Ge-Se bond in 36 (2.2372(5) Å) is slightly longer than Pyykkö’s standard value for a Ge-Se double bond (2.18 Å), and comparable to those of thermodynamically stabilized germaneselones (2.194(1) Å-2.237(2) Å)[17, 33-54]. NBO analyses of 34-36 suggested that the Ge–Ch bond becomes less polarized upon descending group-16 (O: 78.9%; S: 60.5%; Se: 55.3%). Furthermore, unlike the Ge–O bond in 33, the Ge–S and Ge–Se bonds of 35 and 36 exhibit partial double-bond character, as indicated by the calculated WBI values of 1.279 and 1.302, respectively.
-
3.3. Heavier analogues of CO and CO2
Kaupp and Driess have reported the synthesis of bis-NHC-stabilized monomeric silicon chalcogenides. Silicon selenide 38 and diselenide 39 were obtained from the reaction of silylone 37 with elemental selenium[61]. Compounds 38 and 39 serve as base-stabilized heavier analogues of CO and CO₂, respectively. Interestingly, the formation of 38 and 39 is solvent-dependent, i.e., 38 forms in acetonitrile, while 39 forms in THF. Notably, 38 does not react further in acetonitrile with additional Se, while solvent exchange to THF smoothly converts 38 into 39. The 77Se NMR resonances of 38 and 39 appear at –655 ppm and –391 ppm, respectively. The length of the Si–Se bonds in 38 (2.135(1) Å/2.1439(9) Å in the two independent molecules) are consistent with Pyykkö’s standard value for a Si=Se double bond (2.14 Å)[17]. In 39, the longer Si1–Se2 bond (2.241(6) Å) approaches Pyykkö’s standard value for a Si–Se single bond (2.32 Å), while the shorter Si1–Se1 bond (2.129(2) Å) resembles that of 38[17].

Figure 3-4.
a) Synthesis of germaacylium ion 34 and its heavier analogues 35 and 36. b) Crystal structures of 34-36.

Figure 3-5.
a) Synthesis of silicon and selenium analogues of CO (38) and CO2 (39) together with b) their crystal structures.
-
3.4. Heavier analogues of allenes
Sugamata and co-workers have reported on the synthesis of a series of bis(methylene)-λ⁴-chalcogenanes, i.e., 2-chalcogenaallene-type molecules, using bulky silyl substituents[62-65]. The selenium analogue, i.e., bis(methylene)-λ⁴-selane 40, was obtained from the reaction of a transient bis(silyl)carbenoid with elemental selenium. While 40 represents a heavier analogue of a carbonyl ylide, it is also a significant example of a bent-allene–type structure rather than an ylide, being characterized as a symmetric molecule. The 77Se NMR resonance of 40 appears at 1501 ppm, which is characteristic for unsaturated oraganoselenium compounds[66,67]. The crystal structure of 40 exhibits pseudo-C₂ symmetry with a bent C–Se–C configuration. The nearly identical, short C–Se bond lengths (1.801(4) Å/1.808(3) Å) in 40 falls between Pyykkö’s standard values for a C=Se double bond (1.74 Å) and a C–Se single bond (1.91 Å), indicating significant multiple-bond character[17]. An NBO analysis of 40 revealed two C-Se bonds and a 3-center-4-electron π-bond on the C–Se–C moiety.
Bis(methylene)-λ⁴-selane 40 is inert toward a variety of alkynes and alkenes, despite being an analogue of carbonyl ylides, which are known as 1,3-dipole reagents. However, the reaction with hydrogen chloride yielded the corresponding 1,3-adduct (41) in a quantitative yield. Furthermore, treatment of 40 with GeCl₂·dioxane produced unique four-membered ring compound 42, suggesting that the formal [3 + 1] cycloaddition represents a specific reactivity pattern of bis(methylene)-λ⁴-chalcogenanes. The reaction with AuCl·Me₂S selectively afforded the unexpected dinuclear carbene gold(I) complex 43 via the elimination of Ph₂MeSiCl[68].

Figure 3-6.
a) Synthesis of bis(methylene)-λ4-selane 40, and its b) crystal structure.

Figure 3-7.
Reactions of 40.
4. Group-15 – Selenium π-bond compounds

Figure 4-1.
Compounds with terminal E15=Ch bonds.
The synthesis of compounds with multiple bonds between heavier group-15 elements (E15 = P, As, Sb, Bi) and chalcogens (Ch = S, Se, Te) has received increased attention over the last few decades. Phosphine oxides and their heavier analogues, which are P(V) species, are well known as useful reagents in organic synthesis (Figure 4-1). Their heavier analogues should be described as polar single-bonded structures[69]. In contrast, E15(III) chalcogenides are extremely rare species due to their low stability. The stabilization of such compounds typically requires sterically overcrowded ligands to prevent self-oligomerization. Tokitoh and co-workers have reported the selenization of kinetically stabilized diphosphene (G and J; ArP=PAr), distibenes (H and K; ArSb=SbAr) and dibismuthenes (I and L; ArBi=BiAr) to afford the corresponding E15(III) selenides (M-P)[70-72], (Figure 4-2). Nevertheless, evidence for monomeric compounds that bear an unsupported terminal E15(III) chalcogen double bond is still missing. Breunig and co-workers have suggested the presence of a terminal antimony-selenium double bond for tungsten complex Q in benzene solution, albeit that Q forms a dimer in the solid state. In this chapter, notable achievements of E15(III) selenides are described.

Figure 4-2.
Pioneering studies on E15(III) selenides.
-
4.1. Antimony(III) selenides
Dostál and co-workers have reported the synthesis of the first stable antimony(III) selenide using an NCN-pincer ligand[73]. The reaction of the cyclic organoantimony compound of the type Ar4Sb4 (44) with an excess of elemental selenium in THF afforded the corresponding antimony(III) selenide (45) as a highly air-sensitive yellow crystalline solid[74]. The 1H and 13C NMR spectra of 45 displayed only one set of signals, indicating a highly symmetric structure in solution. A single signal at –197 ppm was observed in the 77Se spectrum, and the 77Se CP/MAS NMR spectrum at –153 ppm confirmed that the monomeric structure of 45 in the solid state is retained in solution. In the solid state, 45 is monomeric, with a Sb-Se bond length of 2.4396(7) Å, which is approximately 9% shorter than Pyykkö’s standard value for a Sb–Se single bond (2.56 Å), approaching the length of a Sb=Se double bond (2.40 Å)[17]. The Kohn-Sham orbitals of 45 show that the HOMO corresponds mainly to the selenium lone pair and the HOMO-1 to the Sb-Se π-bond (Figure 4-3). The WBI for Sb-Se in 45 is 1.41. An NBO analysis showed notable charge separation, with NBO charges of +0.999 on Sb and –0.788 on Se. Moreover, the NBO analysis revealed a large back-donation from the lone pair on Se into an empty orbital on Sb, suggesting that the terminal Sb–Se bond in 45 exhibits appreciable double-bond character, albeit that the electron density is strongly polarized toward selenium.
To investigate the impact of donating N atoms on the character of the Sb–Se bond, the same research group synthesized antimony(III) selenides 47[75], 49[76], and 51[77] with different NCN-pincer ligands (Figure 4-4). The reaction of antimony chloride 46 with Li2Se yielded monomeric antimony(III) selenide 47, while 49 and 51 were obtained from the reactions of elemental selenium with stibinidenes 48 and 50, respectively. The 77Se NMR spectra of the antimony(III) selenides showed a single peak at –139.6 ppm (47) and –17.3 ppm (51), which are slightly downfield shifted from that of 45; for 49, no apparent signal was observed. The length of the Sb–Se bonds in 47 (2.4329(5) Å), 49 (2.4497(4) Å), and 51 (2.4371(5) Å) suggest double-bond character similar to that in 45. NBO calculations were performed to analyze the bonding nature of the Sb–Se bond in these antimony(III) selenides, using a consistent level of calculations (Table 4-1). The results indicate that the terminal Sb–Se bonds in these antimony(III) selenides exhibit lower polarity and stronger double-bond character, influenced by the electron-donating strength of the pincer ligands.

Figure 4-3.
a) Synthesis of antimony(III) selenide 45 together with b) its crystal structure. c) Kohn-Sham frontier orbitals of 45.

Figure 4-4.
a) Synthesis and molecular structures of antimony(III) selenides 47, 49, 51.
Table 4-1.
Comparisons among the antimony(III) selenides
45,
47,
49,
51.
|
Sb-Se (Å) |
C-Sb (Å) |
N1-Sb (Å) |
Sb-N2 (Å) |
WBIa)
|
NBO chargea)
|
δ77Se
|
Ref. |
| Sb |
Se |
| 45
|
2.4396(6) |
2.135(4) |
2.461(3) |
2.518(3) |
1.376 |
1.023 |
–0.807 |
–197 |
[74]
|
| 47
|
2.4329(5) |
2.124(3) |
2.395(2) |
2.393(2) |
1.288 |
1.105 |
–0.779 |
–140 |
[75]
|
| 49
|
2.4498(6) |
2.129(1) |
2.549(1) |
2.391(1) |
1.242 |
1.099 |
–0.807 |
— |
[76]
|
| 51
|
2.4371(5) |
2.147(4) |
2.536(3) |
2.458(3) |
1.271 |
1.096 |
–0.811 |
–17.3 |
[77]
|
a) Calculated at the B3PW91-D3(BJ)/SDD level for Sb, and the corresponding 6-31G(d) level for all other atoms.
-
4.2. As(III) selenides
Dostál and co-workers have also reported the synthesis of arsenic(III) selenide 52 using the same NCN-pincer ligand as for the synthesis of 45 (Figure 4-5)[78]. The reaction of a dichloroarsine with Li2Se afforded arsenic(III) selenide 52 as a yellow solid. Its 77Se NMR spectrum showed a resonance at 0 ppm, which is downfield shifted compared to those of other antimony(III) selenides. The terminal As-Se bond (2.2736(5) Å) is significantly shorter than Pyykkö’s standard value for an As–Se single bond (2.37 Å), approaching the length of a typical As=Se double bond (2.21 Å). The NBO charges on As and Se amount to +0.740 and –0.645, respectively. The WBI of 52 (1.48) is higher than those of antimony analogue 45 (1.41).
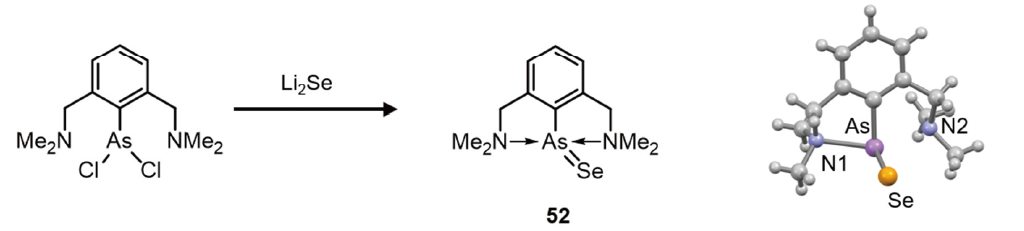
Figure 4-5.
a) Synthesis and molecular structures of arsenic(III) selenide 52.
5. Summary
In this mini-review, we have presented recent examples of isolated compounds that feature both selenium and heavier main-group elements in a π-bond, which are stabilized through kinetic and thermodynamic means using judiciously designed substituents. A notable characteristic of these species is the highly electrophilic nature of the selenium atom in the π-bond, due to the presence of a low-lying π* orbital. This represents a captivating area of study within main-group element chemistry. Despite their remarkable properties, these intriguing compounds remain relatively unexplored, offering significant opportunities for further advancements across various research domains.
Acknowledgments
The authors would like to thank Dr. U.F.J. Mayer from Mayer Scientific Editing (http://www.mayerscientificediting.com/) for his assistance during the preparation of this manuscript. Theoretical calculations in this manuscript have been performed with financial support by JSPS KAKENHI grants 21KK0094 and 23H01943 from MEXT (Japan), and JST CREST grant JPMJCR19R4.
References
- [1] (a) Guo T , Li Z , Bi L , Fan L , Zhang P . Recent advances in organic synthesis applying elemental selenium. Tetrahedron 2022; 112: 132752 (Article number). (b) Sonego JM, de Diego SI, Szajnman SH, Gallo-Rodriguez C, Rodriguez JB. Organoselenium Compounds: Chemistry and Applications in Organic Synthesis. Chem. Eur. J. 2023; 29: e202300030 (Article number). (c) Reich HJ, Hondal RJ. Why Nature Chose Selenium. ACS Chem. Biol. 2016; 11: 821-841. (d) Singh FV, Wirth T. Selenium reagents as catalysts Catal. Sci. Technol. 2019; 9: 1073-1091.
- [2] (a) Ramírez-Gómez A , Gutiérrez-Hernández AI , Alvarado-Castillo MA , Toscano RA , Ortega-Alfaro MC , López-Cortés JG . Selenoamides as powerful scaffold to build imidazo[1,5-a]pyridines using a grinding protocol. J. Organomet. Chem. 2020; 919: 121315 (Article number). (b) Zhao Q, Li G, Nareddy P, Jordan F, Lalancette R, Szostak R, Szostak M. Structures of the Most Twisted Thioamide and Selenoamide: Effect of Higher Chalcogens of Twisted Amides on N-C(X) Resonance. Angew. Chem. Int. Ed. 2022; 61: e202207346 (Article number).
- [3] Wirth T . Organoselenium Chemistry: Synthesis and Reactions Wiley-VCH 2011.
- [4] Pitzer KS . Repulsive Forces in Relation to Bond Energies, Distances and Other Properties. J. Am. Chem. Soc. 1948; 70: 2140-2145.
- [5] West R , Fink MJ , Michl J . Tetramesityldisilene, a Stable Compound Containing a Silicon-Silicon Double-Bond. Science 1981; 214: 1343-1344.
- [6] Yoshifuji M , Shima I , Inamoto N , Hirotsu K , Higuchi T . Synthesis and Structure of Bis(2,4,6-tri-tert-butylphenyl)diphosphene: - Isolation of a True Phosphobenzene. J. Am. Chem. Soc. 1981; 103: 4587-4589.
- [7] Fischer RC , Power PP . π-Bonding and the Lone Pair Effect in Multiple Bonds Involving Heavier Main Group Elements: Developments in the New Millennium. Chem. Rev. 2010; 110: 3877-3923.
- [8] Okazaki R , Tokitoh N . Heavy Ketones, the Heavier Element Congeners of a Ketone. Acc. Chem. Res. 2000; 33: 625-630.
- [9] Suzuki H , Tokitoh N , Okazaki R , Nagase S , Goto M . Synthesis, Structure, and Reactivity of the First Kinetically Stabilized Silanethione. J. Am. Chem. Soc. 1998; 120: 11096-11105.
- [10] NBO calculations were performed using the NBO7 program; for details, see: Glendening ED , Landis CR , Weinhold F . NBO 7.0: New vistas in localized and delocalized chemical bonding theory. J. Comput. Chem. 2019; 40: 2234-2241.
- [11] Pachaly B , West R . Synthesis of a 1,3-Dioxa-2,4-diboretane, an Oxoborane Precursor. J. Am. Chem. Soc. 1985; 107: 2987-2988.
- [12] Tokitoh N , Ito M , Okazaki R . Formation and reactions of a thioxoborane, a novel boron-sulfur double-bond compound. Tetrahedron Lett. 1996; 37: 5145-5148.
- [13] Vidovic D , Moore JA , Jones JN , Cowley AH . Synthesis and Characterization of a Coordinated Oxoborane: Lewis Acid Stabilization of a Boron-oxygen Double bond. J. Am. Chem. Soc. 2005; 127: 4566-4567.
- [14] Franz D , Inoue S . Advances in the development of complexes that contain a group 13 element chalcogen multiple bond. Dalton Trans. 2016; 45: 9385-9397.
- [15] Martínez JP , Trzaskowski B . Structural and Electronic Properties of Boranes Containing Boron-chalcogen Multiple Bonds and Stabilized by Amido Imidazoline-2-imine Ligands. Chem. Eur. J. 2022; 28: e202103997 (Article number).
- [16] Wang H , Zhang J , Hu H , Cui C . Access to B=S and B=Se Double Bonds via Sulfur and Selenium Insertion into a B-H Bond and Hydrogen Migration. J. Am. Chem. Soc. 2010; 132: 10998-10999.
- [17] Pyykkö P , Atsumi M . Molecular Double-bond Covalent Radii for Elements Li–E112. Chem. Eur. J. 2009; 15: 12770-12779.
- [18] Ito M , Tokitoh N , Okazaki R . 1,3,2,4-Diselenastannaboretane, a novel selenium-containing four-membered boracycle: synthesis, structure and thermal cycloreversion into a selenoxoborane. Chem. Commun. 1998: 2495-2496.
- [19] Ito M , Tokitoh N , Okazaki R . Syntheses, structures, and properties of novel four-membered stannacycles, 1,3,2,4-dichalcogenastannaboretanes. Phosphorus Sulfur Silicon Relat. Elem. 1999; 150: 145-148.
- [20] Ito M , Tokitoh N , Kawashima T , Okazaki R . Formation of a borylene by photolysis of an overcrowded bis(methylseleno)borane. Tetrahedron Lett. 1999; 40: 5557-5560.
- [21] Conrad O , Jansen C , Krebs B . Boron-sulfur and Boron-selenium Compounds-from Unique Molecular Structural Principles to Novel Polymeric Materials. Angew. Chem. Int. Ed. 1998; 37: 3208-3218.
- [22] Amii H , Vranicar L , Gornitzka H , Bourissou D , Bertrand G . Radical-type Reactivity of the 1,3-Dibora-2,4-diphosphoniocyclobutane-1,3-diyl. J. Am. Chem. Soc. 2004; 126: 1344-1345.
- [23] Jaiswal K , Prashanth B , Ravi S , Shamasundar KR , Singh S . Reactivity of a dihydroboron species: synthesis of a hydroborenium complex and an expedient entry into stable thioxo- and selenoxo-boranes. Dalton Trans. 2015; 44: 15779-15785.
- [24] Dolati H , Denker L , Trzaskowski B , Frank R . Superseding β-Diketiminato Ligands: An Amido Imidazoline-2-imine Ligand Stabilizes the Exhaustive Series of B=X boranes (X=O, S, Se, Te). Angew. Chem. Int. Ed. 2021; 60: 4633-4639.
- [25] Liu S , Légaré MA , Hofmann A , Braunschweig H . A Boradiselenirane and a Boraditellurirane: Isolable Heavy Analogs of Dioxiranes and Dithiiranes. J. Am. Chem. Soc. 2018; 140: 11223-11226.
- [26] Liu S , Légaré MA , Auerhammer D , Hofmann A , Braunschweig H . The First Boron-tellurium Double Bond: Direct Insertion of Heavy Chalcogens into a Mn=B Double bond. Angew. Chem., Int. Ed. 2017; 56: 15760-15763.
- [27] Zhang X , Liu LL . Crystalline Neutral Aluminum Selenide/Telluride: Isoelectronic Aluminum Analogues of Carbonyls. J. Am. Chem. Soc. 2023; 145: 15729-15734.
- [28] Xu H , Kostenko A , Weetman C , Fujimori S , Inoue S . An Aluminum Telluride with a Terminal Al=Te Bond and its Conversion to an Aluminum Tellurocarbonate by CO2 Reduction. Angew. Chem., Int. Ed. 2023; 62: e202216021 (Article number).
- [29] Anker MD , Coles MP . Isoelectronic Aluminium Analogues of Carbonyl and Dioxirane Moieties. Angew. Chem., Int. Ed. 2019; 58: 13452-13455.
- [30] Back TG , Barton DHR , Britten-Kelly MR , Guziec FS . Synthesis and properties of monomeric selenoketones. J. Chem. Soc. Chem. Commun. 1975; (13): 539.
- [31] Arya P , Boyer J , Carré F , Corriu R , Lanneau G , Lapasset J , Perrot M , Priou C . Formation and Reactivity of Silicon–sulfur and Silicon–selenium Double Bonds. The First X-ray Structure of a Silanethione. Angew. Chem. Int. Ed. 1989; 28: 1016-1018.
- [32] Matsumoto T , Tokitoh N , Okazaki R . Synthesis and Structure of the First Stable Germaneselone. Angew. Chem. Int. Ed. 1994; 33: 2316-2317.
- [33] Kuchta MC , Parkin G . Terminal Sulfido and Selenido Complexes of Tin: Syntheses and Structures of [η4-Me8taa]SnE (E = S, Se). J. Am. Chem. Soc. 1994; 116: 8372-8373.
- [34] Huo SC , Li Y , Zhang DX , Zhou Q , Yang Y , Roesky HW . Synthesis, Characterization, and Reaction of Digermylenes. Chem. Asian J. 2022; 17: e202200141 (Article number).
- [35] Soto-Montero T , Flores-Díaz N , Molina D , Soto-Navarro A , Lizano-Villalobos A , Camacho C , Hagfeldt A , Pineda LW . Dopant-free Hole-transport Materials with Germanium Compounds Bearing Pseudohalide and Chalcogenide Moieties for Perovskite Solar Cells. Inorg. Chem. 2020; 59: 15154-15166.
- [36] Siwatch RK , Nagendran S . Germaester Complexes with a Ge(E)Ot-Bu Moiety (E = S or Se). Organometallics 2012; 31: 3389-3394.
- [37] Pineda LW , Jancik V , Oswald RB , Roesky HW . Preparation of LGe(Se)OH: A Germanium Analogue of a Selenocarboxylic Acid (L = HC[(CMe)(NAr)]2, Ar = 2,6-iPr2C6H3). Organometallics 2006; 25: 2384-2387.
- [38] Harris LM , Tam ECY , Cummins SJW , Coles MP , Fulton JR . The Reactivity of Germanium Phosphanides with Chalcogens. Inorg. Chem. 2017; 56: 3087-3094.
- [39] Siwatch RK , Karwasara S , Sharma MK , Mondal S ; Mukherjee G ; Rajaraman G ; Nagendran S . Reactivity of LGe–NR2 and LGe(E)–NR2 over LGe–Cl and LGe(E)–Cl toward Me3SiX (L = Aminotroponiminate; NR2 = N(SiMe3)2/NC4H4; E = S/Se; X = Br/CN). Organometallics 2016; 35: 429-438.
- [40] Ding Y , Ma Q , Roesky HW , Herbst-Irmer R , Usón I , Noltemeyer M , Schmidt HG . Synthesis, Structures, and Reactivity of Alkylgermanium(II) Compounds Containing a Diketiminato Ligand. Organometallics 2002; 21(24): 5216-5220.
- [41] Tam ECY , Harris LM , Borren ES , Smith JD , Lein M , Coles MP , Fulton JR . Why compete when you can share? Competitive reactivity of germanium and phosphorus with selenium. Chem. Commun. 2013; 49: 10278-10280.
- [42] Foley SR , Bensimon C , Richeson DS . Facile Formation of Rare Terminal Chalcogenido Germanium Complexes with Alkylamidinates as Supporting Ligands. J. Am. Chem. Soc. 1997; 119: 10359-10363.
- [43] Yadav D , Siwatch RK , Mukherjee G , Rajaraman G , Nagendran S . Use of Thio and Seleno Germanones as Ligands: Silver(I) Halide Complexes with Ge=E→Ag-I (E = S, Se) Moieties and Chalcogen-dependent Argentophilic Interaction. Inorg. Chem. 2014; 53: 10054-10059.
- [44] Ding Y , Ma Q , Roesky HW , Usón I , Noltemeyer M , Schmidt HG . Syntheses, structures and properties of [HC(CMeNAr)2Ge(E)X] (Ar = 2,6-iPr2C6H3; E = S, Se; X = F, Cl). Dalton Trans. 2003: 1094-1098.
- [45] Ossig G , Meller A , Brönneke C , Müller O , Schäfer M , Herbst-Irmer R . Bis[(2-pyridyl)bis(trimethylsilyl)methyl-C,N]germanium(II): A Base-stabilized Germylene and the Corresponding Germanethione, Germaneselenone, and Germanetellurone. Organometallics 1997; 16: 2116-2120.
- [46] Karwasara S , Siwatch RK , Jha CK , Nagendran S . Aminotroponiminatosilathio- and Siloxygermylenes: Reactivity Comparison. Organometallics 2015; 34: 3246-3254.
- [47] Karwasara S , Sharma MK , Tripathi R , Nagendran S . Synthesis and Reactivity of N-Aminotroponiminatogermylenepyrrole and its Derivatives. Organometallics 2013; 32: 3830-3836.
- [48] Barman MK , Nembenna S . Mixed guanidinato-amido Ge(IV) and Sn(IV) complexes with Ge=E (E = S, Se) double bond and SnS4, Sn2Se2 rings. RSC Adv. 2016; 6: 338-345.
- [49] Prashanth B , Singh S . Concise access to iminophosphonamide stabilized heteroleptic germylenes: chemical reactivity and structural investigation. Dalton Trans. 2016; 45: 6079-6087.
- [50] Leung WP , Chong KH , Wu YS , So CW , Chan HS , Mak TCW . Synthesis of Chalcogeno[3-(pyrid-2-yl)-1-azaallyl]germanium Complexes. Eur. J. Inorg. Chem. 2006; 2006: 808-812.
- [51] Kocsor TG , Matioszek D , Nemeş G , Castel A , Escudié J , Petrar PM , Saffon N , Haiduc I . Chalcogeno[bis(phosphaalkenyl)] Germanium and Tin Compounds. Inorg. Chem. 2012; 51: 7782-7787.
- [52] Mahawar P , Shukla P , Chandra Joshi P , Singh D , Kumar H , Mukherjee G , Nagendran S . Air and water stable germacarbonyl compounds. Chem. Sci. 2022; 13: 12382-12388.
- [53] Hossain J , Parvin N , Shah BK , Khan S . Four-Coordinate Germylene and Stannylene and their Reactivity towards Se & Te. Z. Anorg. Allg. Chem. 2022; 648: e202200164 (Article number).
- [54] Kim HS , Jung EA , Han SH , Han JH , Park BK , Kim CG , Chung TM . Germanium Compounds Containing Ge=E Double Bonds (E = S, Se, Te) as Single-source Precursors for Germanium Chalcogenide Materials. Inorg. Chem. 2017; 56: 4084-4092.
- [55] Pineda LW , Jancik V , Roesky HW , Herbst-Irmer R . Germacarboxylic Acid: An Organic-acid Analogue Based on a Heavier Group 14 Element. Angew. Chem. Int. Ed. 2004; 116: 5650-5652.
- [56] Ding Y , Ma Q , Roesky HW , Usón I , Noltemeyer M , Schmidt HG . Syntheses, structures and properties of [HC(CMeNAr)2Ge(E)X] (Ar = 2,6-iPr2C6H3; E = S, Se; X = F, Cl). Dalton Trans. 2003; 1094-1098.
- [57] Bordwell FG . Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988; 21: 456-463.
- [58] Tan G , Xiong Y , Inoue S , Enthaler S , Blom B , Epping JD , Driess M . From elusive thio- and selenosilanoic acids to copper(I) complexes with intermolecular Si=E→Cu-O-Si coordination modes (E = S, Se). Chem. Commun. 2013; 49: 5595-5597.
- [59] Xiong Y , Yao S , Driess M . Silicon Analogues of Carboxylic Acids: Synthesis of Isolable Silanoic Acids by Donor-acceptor Stabilization. Angew. Chem. Int. Ed. 2010; 49: 6642-6645.
- [60] Sarkar D , Weetman C , Dutta S , Schubert E , Jandl C , Koley D , Inoue S . N-Heterocyclic Carbene-stabilized Germa-acylium Ion: Reactivity and Utility in Catalytic CO2 Functionalizations. J. Am. Chem. Soc. 2020; 142: 15403–150411.
- [61] Burchert A , Müller R , Yao S , Schattenberg C , Xiong Y , Kaupp M , Driess M . Taming Silicon Congeners of CO and CO2 : Synthesis of Monomeric SiII and SiIV Chalcogenide Complexes. Angew. Chem. Int. Ed. 2017; 56: 6298–6301.
- [62] Sugamata K , Hashizume D , Suzuki Y , Sasamori T , Ishii S . Synthesis and Structure of a Stable Bis(methylene)-λ4-sulfane. Chem. Eur. J. 2018; 24: 6922–6926.
- [63] Sugamata K , Urao Y , Minoura M . A stable bis(methylene)-λ4-selane with a >C=Se=C< bond containing Se(IV). Chem. Commun. 2019; 55: 8254–8257.
- [64] Sugamata K , Asakawa T , Urao Y , Minoura M . Tellurium-Centered Bent Allenes: Synthesis, Characterization, and Reactivity. Inorg. Chem. 2022; 61: 17641–17645.
- [65] Sugamata K , Sasamori T . 2-Heteraallenes. Dalton Trans. 2023; 52: 9882–9892.
- [66] Cullen ER , Guziec FS , Murphy CJ , Wong TC , Andersen KK . Selenium-77 NMR Studies of Some Organoselenium Compounds Containing-selenium Double Bonds. J. Am. Chem. Soc. 1981; 103: 7055–7057.
- [67] Maaninen T , Laitinen R , Chivers T . A monomeric selenium(IV) diimide and a dimeric seleninylamine. Chem. Commun. 2002; 1812–1813.
- [68] Sugamata K , Urao Y , Minoura M . (Thio)(silyl)carbene and (seleno)(silyl)carbene gold(I) complexes from the reaction of bis(methylene)-λ4-sulfane and bis(methylene)-λ4-selane with chloro(dimethylsulfide)gold(I). Dalton Trans. 2020; 49: 7688–7691.
- [69] Heimann S , Bläser D , Wölper C , Haack R , Jansen G , Schulz S . The bonding situation in triethylchalcogenostiboranes - polarized single bonds vs. double bonds. Dalton Trans. 2014; 43: 14772–14777.
- [70] Tokitoh N , Arai Y , Sasamori T , Okazaki R , Nagase S , Uekusa H , Ohashi Y . A Unique Crystalline-state Reaction of an Overcrowded Distibene with Molecular Oxygen: The First Example of a Single Crystal to a Single Crystal Reaction with an External Reagent. J. Am. Chem. Soc. 1998; 120: 433–434.
- [71] Tokitoh N , Sasamori T , Okazaki R . Synthesis of the first stable selenadistibirane and its molecular structure. Chem. Lett. 1998; 27: 725–726.
- [72] Sasamori T , Mieda E , Tokitoh N . Chalcogenation reactions of overcrowded doubly bonded systems between heavier group 15 elements. Bull. Chem. Soc. Jpn. 2007; 80: 2425–2435.
- [73] Dostál L , Jambor R , Růžička A , Lyčka A , Brus J , de Proft F . Synthesis and Structure of Organoantimony(III) Compounds Containing Antimony−Selenium and −Tellurium Terminal Bonds. Organometallics 2008; 27: 6059–6062.
- [74] Dostál L , Jambor R , Růžička A , Holeček J . Syntheses and Structures of Ar3Sb5 and Ar4Sb4 Compounds (Ar = C6H3-2,6-(CH2NMe2)2). Organometallics 2008; 27: 2169–2171.
- [75] Šimon P , Jambor R , Růžička A , Lyčka A , De Proft F , Dostál L . Monomeric organoantimony(III) sulphide and selenide with terminal Sb-E bond (E = S, Se). Synthesis, structure and theoretical consideration. Dalton Trans. 2012; 41: 5140–5143.
- [76] Ganesamoorthy C , Wölper C , Dostál L , Schulz S . Syntheses and structures of N,C,N-stabilized antimony chalcogenides. J. Organomet. Chem. 2017; 845: 38–43.
- [77] Zechovský J , Kertész E , Kerslake V , Hejda M , Mikysek T , Erben M , Růžička, A , Jambor R , Benkő Z , Dostál L . Exploring Differences Between Bis(aldimino)- and Amino-aldimino-N,C,N-pincer-stabilized Pnictinidenes: Limits of Synthesis, Structure, and Reversible Tautomerization-controlled Oxidation. Organometallics 2022; 41: 2535–2550.
- [78] Vrána J , Jambor R , Růžička A , Lyčka A , De Proft F , Dostál L . N→As intramolecularly coordinated organoarsenic(III) chalcogenides: Isolation of terminal As–S and As–Se bonds. J. Organomet. Chem. 2013; 723: 10–14.

 https://ror.org/00hhkn466
https://ror.org/00hhkn466