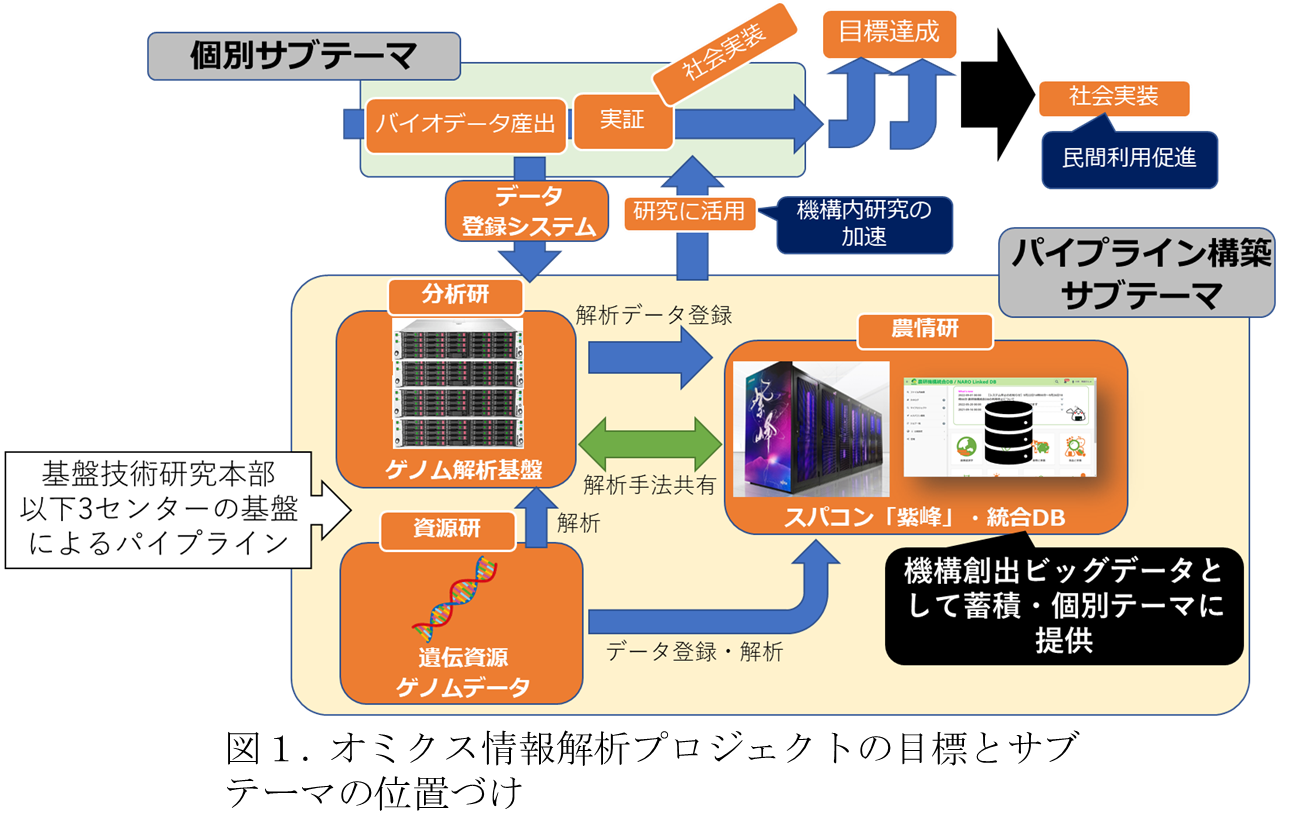
ゲノムデータ連携用メタデータ入出力システム構築
統合 DB には,ファイルの名前や作成者・管理者情報や,対象としている生物種などの,機構内のデータに最低限必要となる一般的なメタデータ項目(以下,NARO Commons)を付与する機能が実装されている.ゲノムデータ連携用のメタデータについても,この NARO Commons と同様に付与することが出来れば,従来の統合 DB へのファイル入出力と同様にゲノムデータを利用できるようになる.これを実現するため,ゲノムデータ連携用のメタデータ入力システムは,この NARO Commons の付与機能を拡張することで実装する.この際,ゲノムデータ以外のオミクス情報のためのパイプライン構築にも拡張可能なように,メタデータを逐次定義し,必要に応じてデータに付与するか否かを選択可能な機能として,カスタムメタデータ付与機能を開発し(以下,カスタムメタ),この機能を用いてゲノムデータ連携用のメタデータを定義した.また,上述の通り,ゲノム解析パイプラインで定義されるメタデータは DRA メタデータに準拠し,このメタデータを DDBJ に登録が可能な形式での出力を行うシステムを構築した.特に,パイプライン構築サブテーマで今後必要となる DDBJ のメタデータ形式は,DRA 以外にも多くのメタデータセットに対応する必要がある(例えば,後述するように gVCF や GWAS 形式など解析データの格納を想定しており,それに対応できるように「Mass Submission System (MSS)」などのメタデータセットの実装を想定している).このため,メタデータ出力システムは,登録されるオミクスデータの種類に即して,個別のメタデータセットを切り替えて DDBJ に登録可能なファイル形式で出力可能なシステムとして構築した.
1.統合 DB カスタムメタ機能と DRA
1) DRA: DDBJ Sequence Read Archive
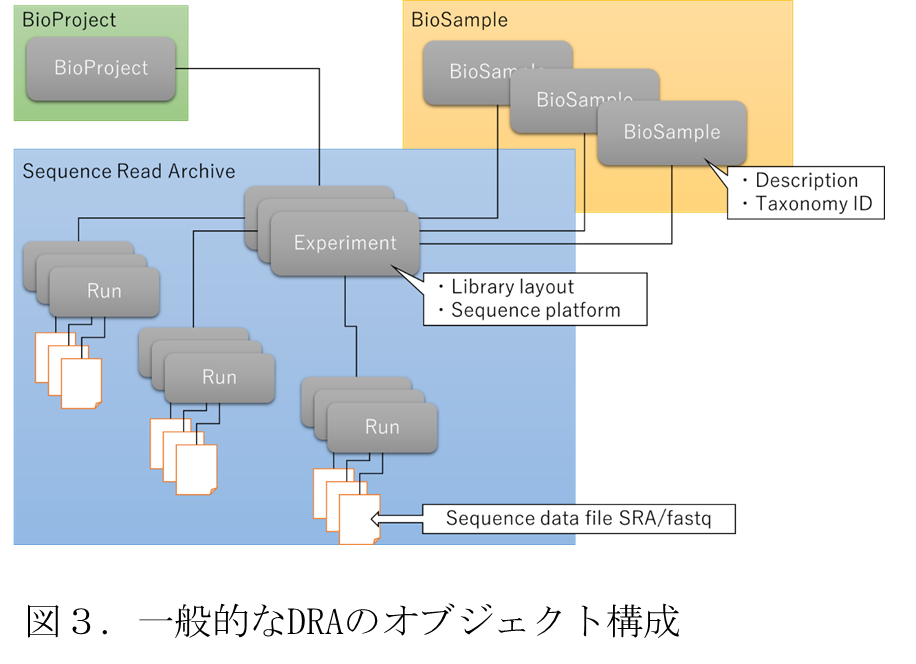
DDBJ センターは,国立遺伝学研究所により運営される,生命科学に関する研究活動をサポートするための機関であり,EBI,NCBI と国際塩基配列データベース連携(INSDC: International Nucleotide Sequence Database Collaboration)を実施している.DDBJ センターは生命科学分野における様々なデータを登録可能なデータベースサービスを提供しており,それらのデータベース毎に,適切にデータを分類・管理するための様々なメタデータが定義されている.ゲノム解析パイプラインでは,これらの内,ゲノムシーケンスを保存するための DRA データベースに関するデータ種別について,それに準拠したカスタムメタデータ及びその入出力機能を統合 DB に実装した.DRA は,生シークエンスデータとアライメント情報を保存するためのデータベースであり,INSDC における NCBI Sequence Read Archive (SRA) 及び EBI Sequence Read Archive (ERA) と共通するメタデータが設定されている.DRA に係る DDBJ 上のメタデータ種別を以下に示す.
・BioProject: 生命科学分野の研究プロジェクトについてのメタデータ.プロジェクトから生み出されるデータを管理する.
・BioSample: 実験データを得るために使用された生物学的な試料及びサンプルについてのメタデータ.
・Experiment: BioSample 1 つと BioProject 1 つを参照し,BioSample から構築した,シークエンス用ライブラリーのメタデータ.
・Run: Experiment に由来するデータファイルをまとめるためのメタデータ.Run に含まれるすべてのファイルは 1 つの SRA/fastq ファイルにマージされる.
・Submission: 登録者,所属組織,公開予定を記載し,同時に登録する DRA オブジェクトをまとめるためのメタデータ.
・Analysis: Run に関連するメタデータで,登録先がないデータを登録する.Analysis は DDBJ/EBI/NCBI で交換されていない.
これらのメタデータ種別の内,Submission は一般的な情報であり,NARO Commons で代替可能である.また,Analysis が付与されるデータは他の DRA に関するデータと結びつかないことから,統合 DB 上にアップロードし,共有すべきデータとしてはあまり適切ではないと考え,統合 DB での Submission 及び Analysis の実装は見合わせた.以上より,ゲノム解析パイプラインでは BioProject,BioSample,Experiment,Run の 4 種類を対象とした.図 3に各メタデータ種別の DRA における基本的な対応関係を示す.
図 3に示すように,4 種のメタデータ種別がそれぞれ Experiment を中心として,それぞれメタデータ項目で対象メタデータを参照する形で運用されている.BioProject 及び BioSample はそれぞれ生命科学研究プロジェクト及び研究で使用される標本を指す独立データベースとなっており,DRA に属するメタデータ種別は正確には Experiment と Run となるが,DRA はいずれかのプロジェクトに属し,なんらかの標本を対象とすることが一般的であるため,図のような対応関係となる.このため,統合 DB についても,DRA 同様に Experiment から BioProject 及び BioSample メタデータを参照可能な形で実装する必要がある.また,DDBJ のデータは全てアクセッション番号と呼ばれる INSDC 間でユニークな ID が付与され,データが一意に識別できるようになっており,Experiment からの参照についても,BioProject や BioSample のアクセッション番号を参照することで実現されている.
統合 DB でこれらの 4 種の DRA メタデータを実装するにあたり,統合 DB ではアクセッション番号を発行できないため,いずれかのデータを参照する方法として,代わりに統合 DB 上での URL を使用することとした.統合 DB の URL はファイルやフォルダ毎に個別に発行されるため,アクセッション番号同様に,統合 DB 上で一意にデータを定めることができる.一方で,BioProject や BioSample については独立したデータベースであることから,既に DDBJ 上に登録されているものを参照したいケースも存在するため,同メタデータ項目には,統合 DB の URL またはアクセッション番号を記載することができるように定義した.ここにアクセッション番号を記載した場合には,統合 DB 上からはその番号の指すデータを直接参照できないものの,後述する DDBJ 登録用のメタデータ出力システムにおいて,出力したファイルを編集せずにそのまま DDBJ に登録することが可能となる.また,ファイル名などの全てのメタデータ種別に共通する項目については,NARO Commons 上の相当する項目を出力し,カスタムメタ上の入力項目から省略した.これは,メタデータ入力時に何度も同様の項目を入力する手間を省くとともに,同様の項目が列挙されることにより入力者が混乱することを避けるためである.
2) 統合 DB DRA カスタムメタ入力システム
DRAの各メタデータ種別は,取り扱うデータに応じてそれぞれ付与されるメタデータが細分化されたメタデータの組(以降,メタデータセット)として定義されている.例えば,BioSample であれば,対象サンプルの生物種や,配列の種類ごとに必要なメタデータ項目が異なるため,それに応じてメタデータセットを切り替える形式となっている.特に,BioSample はサンプルの種別(Sample type)の選択によって大きくメタデータ項目が異なる.そのサンプル種別の中でも,MIxS パッケージ(ゲノム・メタゲノム配列が得られたサンプルのためのパッケージ)と呼ばれる種別を選択した場合にはさらに詳細な分類が表示され,サンプルの生息環境などに応じて分類から選択する必要がある.表1に,対象の各メタデータ種別の内,メタデータ項目を決定する分類項目を示す.
この分類項目ごとに設定されたメタデータ項目は,それぞれ全く異なるわけではなく,一部が共通である場合や,分類によって必須/非必須が切り替わるようになる場合などがあり,DRA と完全に同一の処理をカスタムメタ出力システムに実装する場合には,これらの仕組みを考慮した上で適切に出力内容を切り替える機能が必要となる.しかし,全分類項目の全メタデータ項目ごとに切り分ける設定をカスタムメタ出力システムに実装することは,開発コストが膨大になり困難である.このため,表 1 中の BioSample 選択項目(DNA Data Bank of Japan 2022)から,入出力時に最低限切り分ける必要があるメタデータ項目別にパッケージを書き下し,最終的に以下の分類項目にまとめた.
・BioSample: Generic
・BioSample: Functional genomics
・BioSample Environmental Package: air
・BioSample Environmental Package: host-associated
・BioSample Environmental Package: human-associated
・BioSample Environmental Package: human-gut
・BioSample Environmental Package: human-oral
・BioSample Environmental Package: human-skin
・BioSample Environmental Package: human-vaginal
・BioSample Environmental Package: microbial mat/biofilm
・BioSample Environmental Package: miscellaneous or artificial
・BioSample Environmental Package: plant-associated
・BioSample Environmental Package: sediment
・BioSample Environmental Package: soil
・BioSample Environmental Package: wastewater/sludge
・BioSample Environmental Package: water
構築されたカスタムメタ入力システムのメタデータ設定画面を図 4に示す.ここからカスタムメタデータ種別を入力可と設定することで,図 5に示すように,NARO Commons に加えて設定したカスタムメタ項目の入力欄が追加され,DRA 準拠のメタデータが付与されたデータを保存することが可能となる.この入力欄は,新たに構築されたフォルダ・既存のフォルダまたはファイルに関わらず設定することが可能であり,既存のフォルダ・ファイルに新たに DRA メタデータを付与する,あるいはすでに DRA メタデータが付与されているフォルダから DRA メタデータを削除することが可能である.
3) ゲノムデータの統合 DB への格納
統合 DB 上にカスタムメタデータが適用されたフォルダ・ファイルを格納する方法は 2 通り存在する.一つは,前節で解説したメタデータ入力フォームに値を埋めたフォルダを統合 DB の Web インターフェース上で作成し,そのフォルダにシーケンスデータをアップロードする方法である(以下,ファイル追加機能).もう一つは,カスタムメタデータの項目が埋まったメタデータファイル(エクセル形式)が格納された,シーケンスデータを含むフォルダをアップロードする方法である(以下,一括追加機能).図 3に示したように,DRA では Run メタデータが付与されたフォルダ以下にシーケンスデータが複数含まれる形となるため,実際の運用では,Run に相当するフォルダをファイル追加機能で先に構築するか,一括追加機能でシーケンスデータを含むフォルダのアップロードを行う.アップロード機能は統合 DB 上の汎用の機能であるため,Run に限らず,BioProject,BioSample,Experiment についても同様にファイル追加機能または一括追加機能が利用可能である.ただし,後述するように,フォルダ毎に個別のメタデータが付与された状態でのアップロードに対応できていないため,現在改修を進めている.
なお,統合 DB にシーケンスデータを登録するに際しては,DDBJ では図 3のように,BioProject,Experiment,BioSample などからなるフォルダ階層を事前に構成し,アクセッション番号によってそれぞれ体系づける必要がある.統合 DB 上では,本章 1.1)節で述べたように,代替手段として URL を用いてこれを表現している.特に,Experiment については,Run から必須項目として参照されるほか,BioProject 及び BioSample を必須項目として参照(URL またはアクセッション番号を該当欄に記入)することで,図 3のようなフォルダ階層を実現する.
4) DRA カスタムメタデータ出力システム
統合 DB には,ファイルやフォルダに付与されたメタデータをダウンロードする機能がある.この機能によりダウンロードされるメタデータは,フォルダをアップロードする際に付与することで,メタデータ入力を簡略化するためのものである.この機能では,入力されている値の有無にかかわらず,メタデータ項目すべてをファイルに出力する.このため,この機能で出力したファイルには,DDBJ にデータを登録する際には不要な統合 DB 専用のメタデータ項目が多数存在する.これを DDBJ が受理可能な形式に人手で修正するのは非常に手間がかかり不便であるため,DDBJ が必要とするメタデータ項目だけを出力するシステムを新たに開発し,統合 DB に実装した.
DDBJ では,メタデータ種別が同時に同一データセットに付与されることは無いと考えられるが(BioSample と Experiment が同時に付与されるなど),統合 DB の DRA カスタムメタ入力システムでは,メタデータ種別の排他処理を行っていないため,同時に複数種のメタデータを付与できる.これは,ゲノム解析パイプラインシステムでは DRA のフォルダ構成を統合 DB 上に完全に再現することを目的としているわけではなく,あくまでシーケンスデータや解析データを機構内で容易に連携することを目的としているためである.入力時にこれらのデータについてメタデータ構造を熟慮しなくても保存できるようにしておき,出力時に適切な DRA メタデータ種別でメタデータを出力出来れば,高い利便性を実現した運用が可能となると考えられる.
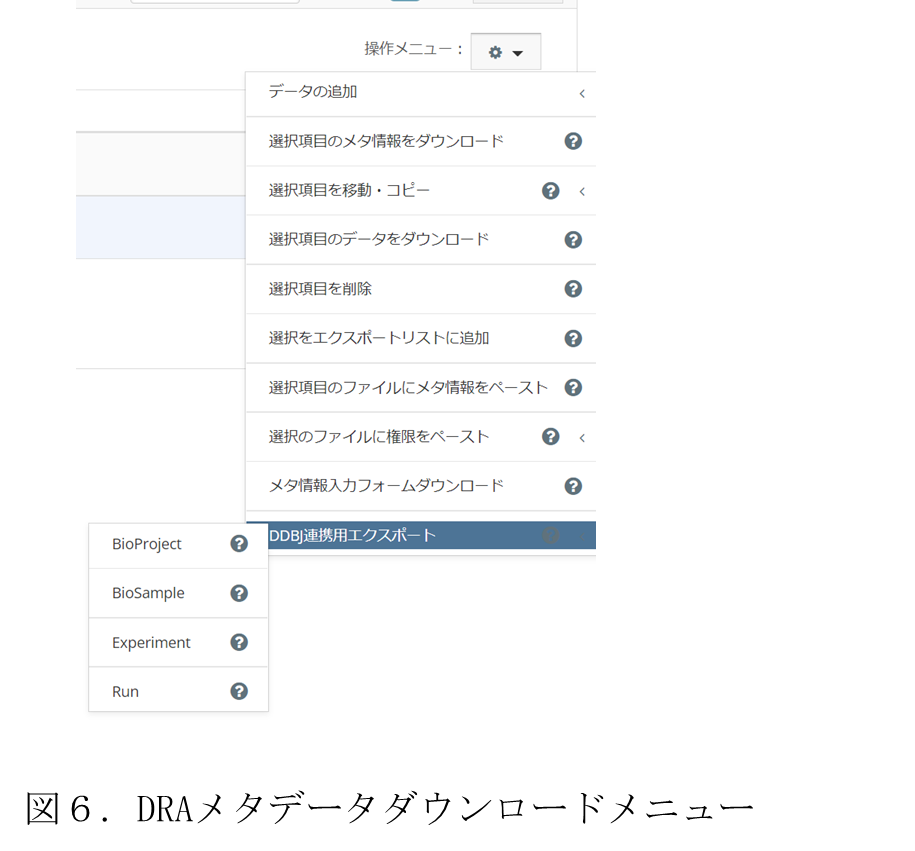
このような背景から,DRA カスタムメタデータ出力システムは,対象フォルダに対して,入力システムで設定されたカスタムメタデータの内,DRA の 4 種メタデータ種別を選択することで,DDBJ に登録したいメタデータを選んで出力可能とした.この選択メニューを図 6に示す.このメニューから,付与されている DDBJ メタデータ種別に対応するものを選択すると,DDBJ に登録可能な形式のメタデータファイル(TSV)形式がダウンロードできる.この際に選択した種別のメタデータが対象フォルダに付与されていない場合はエラーが表示され,ファイルをダウンロードすることができない.また,DDBJ に登録するファイルはアクセッション番号が発行されている場合にはそれを記載する必要があるが,本章 1.4)節 でも述べた通り,統合 DB ではアクセッション番号を発行できないため,TSV ファイルの該当箇所はメタデータ項目に記載が無い限り空欄で出力される.このため,DDBJ に登録するにあたってアクセッション番号が発行済みの場合は手動で入力する必要がある.
メタデータを出力する対象は,画面上に表示されているフォルダ・ファイルに対してそれぞれチェックボックスをチェックすることで選択する.ダウンロードされるメタデータファイルには,チェックされたフォルダ・ファイルのメタデータが列挙される.ただし,フォルダ内のサブフォルダやファイルのメタデータについて,ダウンロード対象の DDBJ メタデータが付与されていたとしても,それらのメタデータを網羅的に取得することはできない.
2.カスタムメタデータ入出力システムを用いたデータ登録の例
農研機構では,ナス科の品種に対する,ナス青枯病抵抗性に関する DNA マーカーの開発を進めており,パイプライン構築とは異なるサブテーマの一つでは,過去のプロジェクトで収集された「世界のナス・コアコレクション」についてのゲノムデータ(以下,ナスコアコレクション)の解析が進められている.このナスコアコレクションについて,既に収集済みであることから,本機能によって統合 DB 上に保存する最初の対象データとして設定されており,現在,このデータの保存を進めている.ナスコアコレクションのデータは 100 サンプルそれぞれについて,次世代シーケンサーで解析された 2 種のシーケンスデータが含まれた Run が含まれている.この Run データの保存作業を進めるにあたり,現状の統合 DB のファイル追加機能及び一括追加機能では,Run ごとに異なるメタデータを付与するには,個別のファイルのメタデータを 1 件ずつ記載するか,あるいは,各サンプル用の統合 DB メタデータファイルを用意する必要がある.これはサンプル数に対して非常に入力コストが高いため,今後ゲノム解析パイプラインから出力される様々なデータの登録を進めるにあたっては,大きな障害となると考えられる.そこで,複数のメタデータを一度に記載した,単一のメタデータファイルをアップロードするだけで,アップロードしたいゲノムデータ全体にメタデータ内容が一度に反映されるよう,カスタムメタデータ入力システムの改修を進めている.
3.DRA 以外の DDBJ データベースメタデータへの対応について
DDBJはバイオデータに対する様々なデータベースサービスを提供しており,それに伴いメタデータ項目もデータベース毎に設定されている.ここまでは,DRA に関するメタデータ付与機能の実装についてのみ解説してきたが,図 2に示すように,本サブテーマでは,ゲノムを解析したデータに対する DDBJ 準拠のメタデータ付与も検討している.現在,構築中のパイプライン上で対象としている解析手法及びデータ形式については後述する.
ここまでに解説したカスタムメタデータ入力システムは,文字通りメタデータをカスタマイズ可能な形で設計しており,これらの DRA 以外のメタデータへの対応は容易に実施できると想定される.
ゲノム解析のためのパイプライン処理プログラム
統合 DB に登録されたゲノムデータを入力として,ゲノム解析パイプライン上で DNA 多型解析やゲノムワイド関連解析(以下,GWAS)を実施できるようにするためのパイプライン処理プログラムについて,構築を進めている.現在はゲノム解析サーバ及び紫峰上で動作するプログラムを構築しており,ゲノム解析パイプラインシステムのための物理ネットワークなどのハードウェア整備が完了次第,パイプラインシステムとしての動作を検証する予定である.
1.DNA多型解析パイプライン処理
次世代シーケンサーで解読された品種・系統等のゲノムデータを参照ゲノム配列にマッピングし,一塩基多型(以下,SNP)や短い挿入・欠失多型(以下,indel)を検出するためのパイプライン処理であり,次の5つの処理で構成されている.
1. 前処理
2. マッピング
3. gVCF ファイルの作成
4. gVCF ファイルの統合および多型サイトの検出
5. 多型サイトのフィルタリング
1.では,次世代シーケンサーで解読された塩基配列(以下,リード配列)中に含まれるアダプター配列や低精度塩基配列の除去を行う.2.では,前処理されたリード配列を参照ゲノム配列にマッピングする.3.では,2.の結果を元に,品種・系統ごとに参照ゲノム配列上の各サイトの遺伝子型等の情報が含まれる gVCF ファイルを作成する.4.では,全ての品種・系統の gVCF を統合して SNP や indel を検出し,5.において様々な統計値を元にフィルタリングを行う.
2.GWAS パイプライン処理
DNA 多型解析パイプライン処理で検出された品種・系統集団の DNA 多型情報を使い,GWAS(ゲノムワイド関連解析,Visscher et al. 2012)を実施するためのパイプライン処理であり,前処理と GWAS 実行の2つの処理で構成される.前処理では,集団の DNA 多型情報が含まれる VCF ファイルに対して,欠損値やマイナーアリル頻度(MAF: Minor Allele Frequency)を元にフィルタリングを行い,GWAS に使用する DNA 多型のセットを抽出する.GWAS を実行するためのツールは数多く公開されており,パイプライン化にあたっては複数のツールを利用できることが重要である.現在,GWAS 用ツールとしての実績が豊富な TASSEL および EMMAX を対象にパイプライン化を進めている.
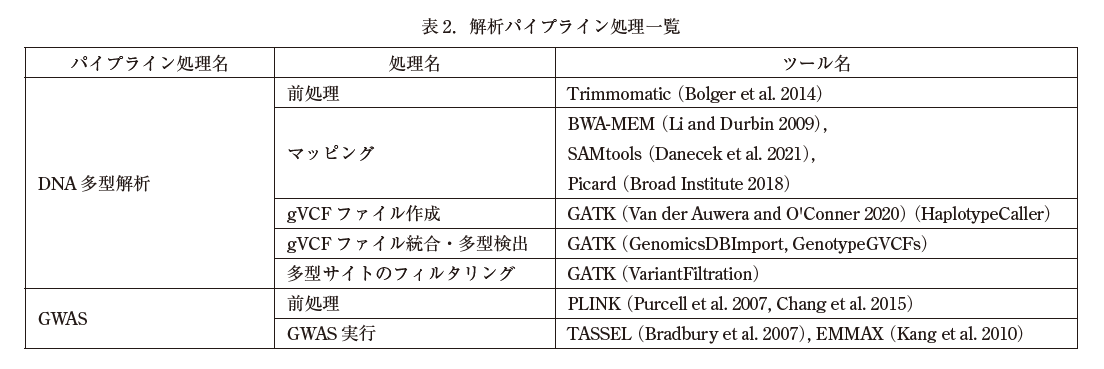
表 2 に,解析パイプライン処理を構成する各処理と組み込んだ各種ツールを示した.各処理を実行するためのシェルスクリプトを構築し,ナスコアコレクションのゲノムデータに適用して動作確認を行った.今後,全ての処理を一気通貫に実行できるようにパイプライン処理として組み上げる作業を進めていく.そして,統合 DB に登録されたゲノムデータを紫峰や分析研のゲノム解析サーバで解析できるように,システム間の連携を進めていく.
3.ゲノム解析パイプライン実現に向けた課題
図 2に示したように,ゲノム解析パイプラインシステムは計算基盤と統合 DB とが連携したハードウェア上でパイプライン処理が実行されることにより実現される.前述したように,紫峰と統合 DB 間は既に連携機能が存在している.この機能を実現するにあたり,紫峰と統合 DB ではユーザ管理が個別に行われており,ユーザ自身がカスタムメタ入出力システムを用いて統合 DB 上にアップロードしたファイルを紫峰上の自身のアカウントから参照することを可能にする必要があった.この課題については,どちらの基盤システムも農研機構の職員管理に使用されている ID をそれぞれベースとしていたことから,この連携の実現は比較的容易であった.
一方で,ゲノム解析サーバのユーザ管理は独立したシステムとなっており,紫峰と同様に統合 DB とを連携することは難しい.これを解決するためには,ゲノム解析サーバのユーザ管理方式を統合 DB 側に合わせるか,あるいはユーザ ID を考慮しない方法などを検討する必要があり,現在議論を進めている.また,ハードウェア的にも,ゲノム解析サーバは分析研の所管である一方,紫峰と統合 DB は農情研の所管であり,設置されているネットワークも異なるため,ゲノム解析サーバと農情研基盤との物理的なネットワークの設置についても検討が必要である.
今後,パイプライン構築サブテーマでは機構内の様々な計算基盤と統合 DB とを連携していくことを想定している.その最初の取り組みであるゲノム解析パイプラインのためのネットワーク設置については,今後のプロジェクト内での拡張を考慮した上でその基礎あるいはモデルケースとして有益な形で実装する必要があるため,慎重に検討を進めている.また,パイプラインによるデータ登録についても,前述したように,実務に当たって大量に登録したいという要求に対しては,メタデータ項目を Web インターフェース上でデータ毎に修正する方法しか現在のところ実装できておらず,一括での入力が可能となるように改修を進めている.