Novel insights into adrenocortical disease through advanced transcriptome analysis methods: focus on single-cell analysis and in silico approaches
2025 Volume 42 Issue 1 Pages 23-29
Details
2025 Volume 42 Issue 1 Pages 23-29
副腎皮質疾患の病態生理の解明は,内分泌分野における重要課題である。近年,シングルセルRNAシーケンシングおよび空間トランスクリプトーム解析の技術革新により,個々の細胞の特性把握や組織内での遺伝子発現の空間的分布を詳細に解析することが可能となった。本稿では,両解析技術の基盤について概説するとともに,大規模データから生物学的な意味を抽出するために必要な,データの前処理から高次解析に至る解析手法について解説する。さらに,両解析技術を用いて明らかになった,副腎皮質の加齢性変化や腫瘍形成過程における新知見を紹介する。シングルセルおよび空間トランスクリプトーム解析により,従来のバルク解析では見出すことができなかった細胞の挙動を捉えることが可能となり,副腎皮質疾患研究に新たな展開をもたらしている。
副腎皮質疾患は,良性腺腫から副腎皮質癌,またホルモン産生異常を伴う機能性腫瘍から非機能性腫瘍まで多岐にわたり,その病態生理の解明は内分泌分野における重要な研究課題である。近年,シングルセルRNAシーケンシング(scRNA-seq)や空間トランスクリプトーム解析といった革新的な技術が登場し,従来のバルク解析では見過ごされていた細胞集団の同定や,組織内での遺伝子発現の空間的分布を詳細に解析することが可能となった。
これらの新規解析技術から得られる大規模データは,適切なバイオインフォマティクス解析により,これまでにない精度で副腎皮質の正常および病的状態を理解することを可能にしている。本稿では,scRNA-seq解析および空間トランスクリプトーム解析の技術的基盤と,それらのデータ解析手法について概説し,これらの技術により明らかになりつつある副腎皮質病変の新たな知見について紹介する。
scRNA-seqは,2009年に最初の報告がなされて以来,急速な技術革新を遂げてきた[1]。現在の技術は,細胞の単離方式によってマイクロウェル方式とドロップレット方式に大別される[2]。マイクロウェル方式では,微細な溝(ウェル)を持つプレート上で個々の細胞を分離し,各ウェル内で細胞の溶解,mRNA捕捉を行う,および細胞識別のためのバーコード配列付加を行う。ICELL8(Takara Bio社)では,ナノディスペンサーを使用して各ウェルに細胞を精密に分注する。BD Rhapsody(BD Biosciences社)では,細胞懸濁液をマイクロウェルカートリッジにロードし,重力による自然落下で各ウェルに細胞を配置する。一方,ドロップレット方式では,マイクロ流体デバイスを用いて,個々の細胞をバーコード配列が付加されたビーズとともに液滴に封入する。液滴内で細胞溶解,mRNA捕捉,細胞識別のバーコード配列付加がなされる(図1)。現在広く用いられている10x Genomics社のChromiumシステムでは,数千から数万個の細胞を一度に解析することが可能である。近年では,ホルマリン固定パラフィン包埋(FFPE)組織からもscRNA-seq解析を行うことができる技術(10x Genomics社のflex kit)が開発され,保存検体を用いた研究への応用が進んでいる。

scRNA-seqにおける細胞分離方式
a:マイクロウェル方式(BD Rhapsody)の概略図。細胞懸濁液をマイクロウェルにロードし,重力による自然落下で細胞を各ウェルに配置。その後,バーコード付きビーズを加えてmRNAを捕捉する。b:ドロップレット方式の概略図。マイクロ流体デバイスを用い,細胞とバーコード付きビーズを液滴内に封入してmRNAを捕捉する。
空間トランスクリプトーム解析技術は,組織における遺伝子発現の空間的分布を解析する手法であり,主にシーケンスベースとイメージングベースの2つのアプローチが存在する(図2)。シーケンスベースの代表例である10x Genomics社のVisiumでは,6.5mm四方のキャプチャーエリアに直径55μmのスポットが約5,000個配置された専用のスライドガラスを使用する。各スポットでは,凍結組織の場合は組織から遊離したmRNAを直接キャプチャーし,FFPE組織の場合は特異的なハイブリダイゼーションプローブを用いてmRNA断片から遺伝子発現情報を取得する。また,各スポットには位置情報を識別できるバーコード配列が付加されており,遺伝子発現情報に位置情報バーコードを付加してライブラリを調製し,シーケンスすることで,組織上の特定の位置における遺伝子発現を網羅的に解析することが可能となる。従来のVisium技術では空間分解能が5~10細胞/スポットに限られていたが,新しいプラットフォーム(Visium HD)では,キャプチャーエリア内に数百万のスポットが隙間なく並んでおり,1細胞以下(サブセルラー)の解像度での解析が可能である。ただし,スポット毎の遺伝子発現であり,細胞単位ではないことに注意しなければならない。一方,イメージングベースの手法(10x Genomics社のXenium,Vizgen社のMERSCOPE,NanoString社のCosMxなど)では,組織切片上でin situハイブリダイゼーションにより直接mRNAを検出し,サブセルラーレベルの解像度でその位置情報を保持したまま発現量を定量する。従来のイメージングベース解析では解析可能な遺伝子数は数百程度と制限があったが,最新の技術では数千遺伝子レベルでの発現解析が可能となっている。シーケンスベースとイメージングベースともに新鮮凍結,FFPE組織のいずれにも適用可能であり,多様な研究目的に応じた解析が可能である。
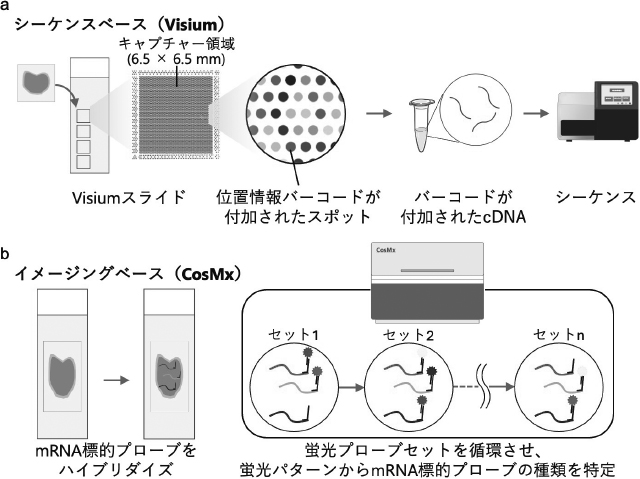
空間トランスクリプトーム解析
a:シーケンスベース解析(Visium)の概略図。スライド上のキャプチャー領域には,mRNAを捕捉するスポットが配置されている。各スポットには位置情報を示すバーコードが付加されており,捕捉されたmRNAはバーコードとともにライブラリ化され,次世代シーケンサーを用いてシーケンスされる。これにより,遺伝子発現情報が位置情報と関連付けられる。b:イメージングベース解析(CosMx)の概略図。mRNA標識プローブを組織内のmRNAにハイブリダイズさせた後,mRNA標識プローブに対応する蛍光プローブセットを用いて蛍光パターンを繰り返し撮影し,プローブの種類を特定する。これにより,位置情報を伴った遺伝子発現情報が得られる。
scRNA-seqおよび空間トランスクリプトーム解析から得られるデータは,数万個の細胞や数千個のスポットにおける数万種類の遺伝子発現情報を含む大規模なデータセットである。各プラットフォームには,遺伝子発現量データの可視化(次元削減)や細胞種分類(クラスタリング),発現変動解析を行うための標準的解析ソフトウェアが提供されている。しかし,より高度な解析として遺伝子発現データと数理的手法を組み合わせた解析(軌道解析や細胞間相互作用解析など)を行うためには,RやPython言語で作成された専用のパッケージを用いる必要がある。R言語ではSeurat,Python言語ではScanpy/Squidpyが広く用いられており,それらを基盤に高次解析のパッケージが展開されている[3~5]。なお,空間トランスクリプトームデータの解析は,現状ではscRNA-seq解析ツールを基本としており,本稿では主にscRNA-seq解析について詳述する。
scRNA-seqのデータ解析は大きく,(1)データの前処理,(2)基本解析,(3)高次解析の3つのステップで行われる(図3)。第一のステップである前処理では,データの品質管理を行う。総リード数,検出遺伝子数,ミトコンドリア遺伝子の割合に基づく低品質細胞の除外を行う。ドロップレット型プラットフォームから得たデータの場合,ambient RNA(細胞が壊れることで溶液中に放出されたRNA)により生じるバックグラウンドノイズを除去するため,CellBenderやSoupXなどのツールを用いる[6,7]。また,2つ以上の細胞が1つの液滴に取り込まれたダブレットの検出と除去には,DoubletFinderやScrubletなどのツールが利用される[8,9]。その後,細胞間のシーケンス深度の差を補正するためデータの正規化を行う。また,異なる実験バッチ間では,実験条件やシーケンス深度の違いにより系統的な変動(バッチエフェクト)が生じるため,複数のサンプルを統合して解析する際にはバッチエフェクトの補正が必要となる。この補正には,HarmonyやscVIなどが用いられる[10,11]。

scRNA-seqデータ解析フロー
scRNA-seqデータ解析では,まず遺伝子発現データの品質管理(低品質細胞のフィルタリングやノイズ補正)と正規化を行う。その後,次元削減やクラスタリングで細胞集団を分類し,細胞種を同定する。最後に,軌道解析や細胞間相互作用解析を用いて,細胞状態の変化や相互作用を推定し,生物学的解釈を行う。
第二のステップである基本解析では細胞種の同定を目指す。数万遺伝子からなる高次元の発現データを,主成分分析により20~50次元程度まで次元を削減した後,非線形次元削減手法であるUMAPやtSNEを用いて2次元あるいは3次元に圧縮し可視化する。続いて教師なし学習によるクラスタリングを行い,細胞を分類する。代表的な手法としてLouvain法やLeiden法があり,これらは細胞間の類似度に基づいてグラフを構築し,クラスターを同定する。各クラスターについて,統計的手法により特徴を示すマーカー遺伝子を抽出し,既知の細胞種特異的マーカーとの比較により細胞種を同定する。参照データセットを利用した細胞種の自動アノテーションツールも存在し,これらのツールは,大規模な参照アトラスや学習済みモデルを活用し,新規データに対して高精度な細胞型の推定を可能にする。SingleRは,参照データセットの遺伝子発現プロファイルとの相関を計算し,各細胞に最も類似した細胞種を割り当てる手法を採用している[12]。CellTypistは,ロジスティック回帰モデルを基盤とした軽量な細胞型予測モデルを提供している。このモデルは大規模な参照データセットを学習して構築されており,新規データに対して高速かつ正確な細胞型の注釈を可能にする[13]。
第三のステップである高次解析では,より詳細な生物学的解釈を目指す。擬似時間解析やRNA velocityなどの軌道解析では細胞状態の変化を推定する。擬似時間解析ツール(Monocle3,Slingshotなど)では,遺伝子発現パターンの類似性に基づいて細胞状態を順序付けし,細胞状態変化の軌跡を推定する[14,15]。一方,RNA velocity解析ツール(velocyto,scVeloなど)では,スプライシング前後のmRNA量の比から細胞状態の変化の方向性を予測する[16,17]。遺伝子制御ネットワーク(GRN)解析ツール(SCENIC,CellOracleなど)では,転写因子とその標的遺伝子の関係性から細胞状態制御の分子メカニズムを推定する[18,19]。SCENICでは,転写因子と標的遺伝子の共発現解析およびモチーフ解析により遺伝子制御ネットワークを推定する。CellOracleでは,scRNA-seqデータとATAC-seqデータ,またはプリセットされた転写因子-ターゲット遺伝子データを統合し,機械学習アプローチでGRNを構築する。構築されたGRNを用いて,特定の転写因子の発現変化(ノックアウトまたは過剰発現)が細胞状態に与える影響をin silicoでシミュレーションし,細胞分化や転写因子の機能を予測することができる。細胞間相互作用解析では,既知のリガンド-受容体ペアのデータベースを利用したツール(CellChat,CellPhoneDBなど)により,細胞間の情報伝達を網羅的に推定する[20,21]。
scRNA-seqデータと空間トランスクリプトームデータの統合解析も重要である。例えば,10x Genomics Visiumの場合,1スポットに複数の細胞が含まれることから,scRNA-seqデータから得られた細胞型情報を用いて,各スポットにおける細胞型の構成を推定する(Deconvolution解析)ことで,組織内の細胞分布をより詳細に理解することが可能となる。10x Genomics Xeniumのような空間解像度が細胞単位またはサブセル単位に対応する技術では,scRNA-seqデータで得られる遺伝子発現の網羅的な情報と統合することで細胞間相互作用や微小環境の空間的な特徴を高精度で解析できる。
副腎皮質は,アルドステロンを産生する球状層,コルチゾールを産生する束状層,副腎アンドロゲンを産生する網状層からなる特徴的な層構造を有する。生理的および病的状態における副腎皮質の変化を理解する上で,細胞の不均一性を単一細胞レベルで捉えつつ,それらの空間的な分布や層構造との関係性を直接的に解析できるscRNA-seq・空間トランスクリプトーム解析は特に有用である。われわれは,これら最新の解析手法により,従来のバルク解析では見出すことができなかった,副腎皮質の層構造特異的な変化や細胞分化の過程,さらには病変における微小環境の役割について解析を行ってきた。以下にその研究成果の概要を述べる。
副腎皮質は加齢に伴い,球状層が断続的となり,束状層が拡大するが,この加齢性変化の分子メカニズムは不明であった。加齢副腎のscRNA-seq・空間トランスクリプトーム解析により,加齢に伴い副腎皮質において炎症反応と細胞老化が増加し,特に束状層において顕著であることが明らかとなった[22]。さらに,GRNおよびin silico摂動解析により,activator protein 1(AP-1)の活性化が束状層細胞においてその遺伝子発現変化を誘導し,それによって束状層細胞から網状層細胞への分化を阻害することが示唆された(図4a)。

副腎皮質における加齢性変化や腫瘍前駆病変の解析
a:加齢ヒト副腎皮質のトランスクリプトーム解析。(左図)空間トランスクリプトーム解析により,加齢に伴い球状層(ZG)が断続的となり,束状層(ZF)が拡大する変化が確認された。(右図)同一組織ブロックのscRNA-seq解析から,高齢副腎皮質のZF細胞でJUN(activator protein 1の構成遺伝子)の活性化を同定。これを若齢副腎皮質データに対してin silico摂動解析で過剰発現させた結果,ZFから網状層(ZR)細胞への分化抑制が示唆された。b:アルドステロン産生細胞クラスター(APCC)のRNA velocity解析により,一部のZG細胞からAPCCへの分化が推定された。c:コルチゾール産生腺腫(CPA)に付随するsteroids-producing nodule(SPN)の空間トランスクリプトーム解析。ZF-likeとZR-likeの2層構造を呈し,異なる遺伝子発現プロファイルを有することが示された。文献[22~24]より引用し一部改変。
副腎皮質腫瘍の形成過程についてもscRNA-seqと空間トランスクリプトーム解析により,新たな知見が得られている。アルドステロン産生腺腫(APA)の前駆病変と推定されるアルドステロン産生細胞クラスター(APCC)は,副腎皮質被膜下・球状層領域に観察される直径1mm前後の微小な病変であり,これまでのバルク解析では詳細な解析が困難であった。scRNA-seq解析により,APCCはCYP11B2陽性球状層細胞とは異なる遺伝子発現プロファイルを有していることが示された[23]。RNA velocity解析の結果,球状層細胞の一部がAPCCへと進展することが示唆された(図4b)。さらに,コルチゾール産生腺腫(CPA)の前駆病変と推定されるステロイド産生結節(steroids-producing nodule:SPN)を同定し,その空間トランスクリプトーム解析を行った(図4c)。SPNは束状層様構造と網状層様構造からなり,それぞれ特徴的な遺伝子発現プロファイルを有しており,束状層様構造においてWNT/βカテニンシグナルが活性化しており,擬似時間解析により束状層様構造の拡大がCPAの形成につながることが推定された[24]。
これらの病変形成過程において,微小環境,特にマクロファージが重要な役割を果たすことが示された。加齢による副腎皮質の変化では,マクロファージを介した炎症反応と細胞老化の誘導が重要なメカニズムとして同定された。同様に,SPNの網状層様構造においては,マクロファージの集積が観察され,これらが腫瘍進展を抑制する方向に働いている可能性が示唆された。これらの知見は,副腎皮質の病変形成過程における微小環境の多様な役割を示している[22,24]。
このように,scRNA-seq・空間トランスクリプトーム解析の導入により副腎皮質病変の分子メカニズム解明が飛躍的に進歩している。
本稿では,scRNA-seq・空間トランスクリプトーム解析について,データ解析の実際や,副腎皮質研究における応用事例を交え概説した。いずれの解析も,研究用に取得保存が必要だった凍結組織ではなく,一般的な診療過程で得られるFFPE組織を材料に行うことができ,以前よりもサンプル収集のハードルが下がっている。加えて,イメージングベースの空間トランスクリプトーム解析技術の進歩は目覚ましく,近い将来2万種類弱の遺伝子に対応し,FFPE切片からサブセルラーレベルの空間情報を保持した網羅的遺伝子発現を捉えることが可能になる。今後のデータ集積により,副腎皮質疾患の分子病態のより深い理解につながることが期待される。