Abstract
The mass fraction of each crystalline phase in inorganic materials can be investigated using the Rietveld refinement of the X-ray diffraction (XRD) patterns. For quantitative analysis, differences in the values of the linear absorption coefficient, μ, among the crystalline phases must be considered when certain X-ray sources are used, because such differences often affect their mass fractions. Herein, we evaluate the effects of the differences between the Cu and Co Kα X-rays on the mass fractions of the crystalline phases in iron sintered ores using the XRD-Rietveld method by performing two types of XRD measurements. Type 1 samples modeled materials with two different particle size combinations of α-Fe2O3 and ZnO. Type 2 samples used powder mixtures to simulate iron sintered ores composed of α-Fe2O3, and synthesized SFCA and SFCA-I in various mass fractions. Moreover, a correction method was developed using the Taylor-Matulis (TM) correction that considers the μ of each phase and the average particle diameter of each crystalline phase determined by scanning electron microscopy with energy dispersive spectroscopy. For type 1 samples, results that were in good agreement with the initially-charged mass fractions could be obtained using the TM correction, even in the presence of significant differences in R between α-Fe2O3 and ZnO. The results for type 2 samples confirmed that quantitatively accurate mass fractions could be obtained using the TM correction with an accuracy of approximately ±3 mass% for Cu and Co sources, whereas the error was greater than ±3 mass% for Cu source when the TM correction was not applied.
1. 緒言
アジア太平洋地域において,高炉に挿入される鉄源の約80 mass%は焼結鉱である。焼結鉱は主に,α-Fe2O3,Fe3O4,および多成分カルシウムフェライト(silico-ferrites of calcium and aluminum (SFCA1); Ca2(Ca,Fe,Al)6 (Fe,Al,Si)6O20,SFCA-I2); (Ca,Fe)4(Fe,Al)16O28)の結晶相で構成されている。その他に,10 mass%未満のケイ酸塩系のスラグや非晶質も含まれると考えられている。焼結鉱中の各相の種類や量は,強度や被還元性などの焼結鉱の特性を制御する上で重要な因子の一つである3,4)。焼結鉱中の各相の質量分率の定量には,一般に光学顕微鏡画像から得られる各結晶相組織の面積比が利用されている5)。加えて,電子プローブマイクロアナライザーまたは走査型電子顕微鏡法-エネルギー分散型分光法(Scanning electron microscopy-Energy dispersive X-ray spectroscopy: SEM-EDS)のような元素分析手法も,焼結鉱中の各相の質量分率の定量に用いられている6,7)。しかし,密度や化学組成が類似しているSFCA相とSFCA-I相をこれらの方法で同定するのは難しい。さらに,実プロセスのような非平衡プロセスで生成した場合のこれらの相の化学組成や固溶範囲は,明らかになっておらず,成分からの定量が困難となる。
X線回折(XRD)-Rietveld法8)は,結晶相の結晶構造の解析や質量分率の定量に広く用いられている9)。Rietveld法は,結晶の単位格子の種類,各サイトの原子種や原子座標などの各結晶相の結晶学的パラメーターに基づいて計算した回折図形と実験で得られたそれらの差分を非線形最小二乗法を用いて最小になるようにして,各結晶相のそれらのパラメーターの精密化や質量分率の定量を行う手法である8,9)。X線および中性子回折図形のRietveld法のデータ解析には,フリーソフト(RIETAN-FP10),Profex11),MAUD12))や市販のソフトウェア(PDXL2,HighScore13),Siroquant14))が広く用いられている。
これまで,焼結鉱中の結晶相の定量にXRD-Rietveld法を適用した例は次の文献15,16)等で報告されている。しかし,XRD-Rietveld法では,試料とX線源の組み合わせによっては,各結晶相の質量分率を正確に行えない場合があり,例えば以下の様な場合には注意が必要である。(i)鉱石の主成分であるFeは,実験室系のXRDのX線源として広く利用されるCu Kα線を強く吸収する。(ii)XRD測定に供する焼結鉱の粉末試料中の各結晶相の粒子径分布が広範囲にわたる。こうした場合には,XRD-Rietveld法を用いて結晶相の質量分率を定量する際,マイクロアブソープション(MA)効果17)と呼ばれるX線の吸収効果を考慮する必要がある。
MA効果とは,XRD測定の際に,X線の一部が試料に吸収される18)ために,測定される回折強度が変化する現象で,質量分率の誤差の原因のひとつになる。この吸収の度合いを示す線吸収係数μは質量吸収係数μmと密度ρの積で表される。μは,各結晶相の組成と入射X線の波長(エネルギー)によって決まる。さらに,複数の結晶相を含む試料の場合,各結晶相のX線回折強度は,それぞれの粒子径分布や粉末試料の試料ホルダーへの充填率によっても変化する。こうしたMA効果に対して質量分率の定量値を補正する方法として,Brindleyによって提案された補正法19)やそれを改良した補正方法(Taylor-Matulis(TM)補正法20))があり,無機物質の結晶相質量分率の定量確度の向上のために使われている。MA効果の詳細な議論や適用の報告は次の文献21-24)に詳しい。
XRD-Rietveld法が焼結鉱に適用された例15,16)は確かにあるが,焼結鉱中の結晶相の定量に対するMA効果の程度やTM法を用いたそれらの値の補正効果を報告した研究はない。焼結鉱には,SFCAやSFCA-Iの様に類似の結晶構造を持つ複数の結晶相や結晶性の低い相が含まれている。これらは,XRD-Rietveld法による質量分率の確度やそれらのTM補正法の効果にも影響を与える可能性がある。Pedersonらは市販の試薬を用いて,Brindley補正法の有用性を報告している25)。ただし,製鉄原料については,粉末試料の調整時に結晶相や非晶質相毎に粉砕の程度が異なったり,同一の結晶相であってもその化学組成に幅があったりするため,線吸収係数と各結晶相の粒子径分布を個別に評価することは難しい。したがって,MA効果の補正係数を試料中の各結晶相について個別に算出することは一般に困難である。焼結鉱の定量において,鉄鉱石の主要結晶相であるα-Fe2O3やFe3O4が強くX線(特にCu Kα)を吸収することに加え,結晶相間の硬度の差異により各結晶相の粒子径分布が異なる可能性があるため,MA効果の影響は大きいと想定される。したがって,製銑工程において主要な高炉鉄源のひとつである焼結鉱のXRD-Rietveld法による結晶相の定量について,MA効果の程度とその補正法20)の有用性を調査することが重要であると考えた。
実験室系でのXRD測定において,X線源としては,Cu KαまたはCo Kα線源(Kα1エネルギーがそれぞれ8.048と6.924 keV)が一般的に用いられることが多い。Cu Kα線を使用した場合,Feの吸収係数はFeのK吸収端(7.120 keV)の近傍で大きくなるため,Feを多く含有する結晶相の回折図形では,その結晶相は実際の仕込みよりも質量分率が小さく見える回折図形が得られる。試料に含まれる各結晶相の質量分率を正確に定量するためには,上記に述べたMA効果を考慮して,X線源と試料中の結晶相の組み合わせを検討する必要がある。一方で,Cu Kα線源は長寿命,低コスト,高出力であることから,古くから広く使用されてきた。そのため,焼結鉱にFeを多く含有する結晶相が含まれていても,Cu Kα線を用いてXRD測定を行わなければならない場合がある。言い換えると,工業的には,工場内で利用できる機器の制約により,仮に試料との相性が悪い場合でも特定のX線源を使用せざるを得ない場合があると考えられる。
本研究では,XRD-Rietveld法による焼結鉱中の結晶相の質量分率の定量確度を向上させるために,2種類の試料系についてTM補正法の効果を検証した。第1の試料系(Type 1)は,粒径が異なるα-Fe2O3とZnOの2種類の混合試料(どちらも質量比50%/50%)である。各試料で,各結晶相の質量分率をXRD-Rietveld法で定量し,線吸収係数と平均粒径を考慮したTM補正法の効果を検証した。第2の試料系(Type 2)として,焼結鉱を模擬しα-Fe2O3と合成したSFCAおよびSFCA-Iの混合物試料について検討した。実験室系におけるXRDの一般的なX線源であるCu Kα(MA効果が大きい)とCo Kα(MA効果が小さい)が,定量値に与える影響を調査するために,6種の試料のXRD測定を行い各結晶相の質量分率を求めた。Type 1と同様に,線吸収係数と平均粒径を考慮して,各定量値に対するTM補正法の効果も検証した。ここで,Type 2については,SEM-EDSで計測したα-Fe2O3と多成分カルシウムフェライト(SFCA+SFCA-I)の粒径分布から各結晶相の平均粒径を算出し,TM補正に用いた。
2. 実験
2・1 試料作製本研究では,MA効果が大きい場合に,TM補正法を用いてXRD-Rietveld法における質量分率の定量値の確度を改善することが可能か否かを検証するため,(Type 1)α-Fe2O3とZnOの粒子径の異なる混合試料と(Type 2)焼結鉱を模擬したα-Fe2O3,SFCA,SFCA-Iから成る混合試料を作製した。
平均粒径の異なるα-Fe2O3とZnOの混合試料(Type 1)を調整し,2種類の平均粒径の組み合わせにおいて,Cu Kα線における線形吸収係数が大きい結晶相(α-Fe2O3)と,小さい結晶相(ZnO)の混合試料におけるMA効果とそのTM補正法の効果を検証した。試料は平均粒径が0.5 μmまたは2 μmのα-Fe2O3と0.6 μmのZnO粉末を同一質量分率(50%/ 50%)で混合した2種類を調整した。α-Fe2O3(高純度化学研究所,4 N,0.5 μm)またはα-Fe2O3(高純度化学研究所,4 N)の2 mm粒子を粉砕して得た平均粒径が20 μm(レーザー散乱法により測定)のα-Fe2O3をZnO(高純度化学研究所,4 N,0.6 μm)と同一質量(50%/50%)で混合した。
焼結鉱中の主要相である3種の結晶相(α-Fe2O3,SFCA,SFCA-I)を含んだ焼結鉱を模擬した試料(Type 2)を用いて,TM補正法の適用前後の各結晶相の質量分率を比較検証した。試料は,α-Fe2O3,SFCA,SFCA-Iを含む異なる質量分率比の6つの試料を調整した。SFCAとSFCA-Iの単相試料は,Hamiltonら1)とMummeら2)が報告した手順に従って粉末焼結法で合成した。それらの合成には,α-Fe2O3(高純度化学研究所,4 N,0.5 μm),CaCO3(関東化学,4 N,12 μm),α-SiO2(高純度化学研究所,99%,1 μm),α-Al2O3(高純度化学研究所,4 N,1 μm),およびMgO(高純度化学研究所(4 N,1 μm)の試薬を出発物質として用いた。Table 1に示す組成比率で調整した粉末混合物(約3 g)を34.5 kNの圧力で直径13 mmのタブレット(2,3個)に成形し,まず脱炭酸を目的に800°Cで仮焼きした。本焼成は,1200°C(SFCA)と1230°C(SFCA-I)でそれぞれ約60時間行った。均質化のため,粉砕・成型・熱処理を3回繰り返した。合成したSFCAとSFCA-IのXRDパターンをFig.1に示す。どちらのXRDパターンにも不純物としてα-Fe2O3が含まれているが,ピーク強度からそれぞれ99%と95%以上の単相が得られたと考えられる。α-Fe2O3と合成したSFCA,SFCA-IをTable 2に示す質量分率で混合し,モデル試料を調整した。α-Fe2O3は,平均粒径が約2 mmの試薬(高純度化学研究所,4 N)を粉砕して調整した。メノウ乳鉢と乳棒を使用し,約30分間,各モデル試料を粉砕した。
Table 1. Initial composition of SFCA and SFCA-I (mass%).
| CaCO3 | α-Fe2O3 | α-Al2O3 | α-SiO2 | MgO |
|---|
| SFCA 1) | 25.7 | 59.1 | 5.3 | 7.8 | 2.1 |
| SFCA-I 2) | 18.2 | 76.9 | 4.9 | − | − |
Table 2. Mass fractions of model samples (mass%).
| No. | α-Fe2O3 | SFCA | SFCA-I |
|---|
| 1 | 50.0 | 25.0 | 25.0 |
| 2 | 49.8 | 12.6 | 37.6 |
| 3 | 49.8 | 37.5 | 12.7 |
| 4 | 50.0 | 33.4 | 16.6 |
| 5 | 50.0 | 39.8 | 10.3 |
| 6 | 49.9 | 45.1 | 5.0 |
MA効果が大きい場合でも,TM補正法を用いてXRD-Rietveld法で算出した定量値が補正できるか検証するため,(Type 1)平均粒径が異なる2種類のα-Fe2O3とZnOの混合試料と(Type 2)6種の模擬焼結鉱試料について,(Type 1)はCu KαX線源を,(Type 2)はCu KαとCo KαX線源を用いてXRD測定を行った。XRDパターンをRietveld法で解析して,結晶相の質量分率を算出した。さらに,各結晶相の線吸収係数と平均粒径を考慮したTM補正法20)を適用し各質量分率の補正を試みた。Type 2の試料では,SEM-EDSの自動粒子解析機能を用いて各結晶相の平均粒径を算出し補正に用いた。
XRD測定にはディフラクトメーター(SmartLab®(リガク))を使用した。試料ホルダーの大きさはφ20 mm×深さ0.2 mmである。管電流と管電圧は,Cu KαとCo Kαで,それぞれ40 mA/40 kV,36 mA/40 kVに設定した。ゴニオメーターの半径は285 mmである。X線検出器は高速1次元検出器(D/teX(リガク))を使用した。測定にはBragg-Brentano集光系を採用し,Cu KαとCo KαでそれぞれNi,Feフィルターを使用した(Kβフィルター法)。測定条件は次の通りであった。測定角度2θ=20~80°,ステップ角幅Δ2θ= 0.02°,スキャン速度=1°/min,試料面法線周りの回転数= 60 rpm。発散スリット(DS)は1/2°,受光スリットRS1とRS2の幅はそれぞれ8 mmと13 mmとした。
XRD-Rietveld法による解析には,PDXLソフトウェア(version 2.8.1.1,リガク)を使用した。解析に使用した各結晶相の結晶構造パラメーターは次のとおりである。α-Fe2O3,ZnOは,ICDDカード番号01-080-237726),01-079-020627)のデータを用いた。SFCAはHamiltonら1)が,SFCA-IはMummeら2)が報告したパラメーターを用いた。解析では,原子座標と温度因子は初期値に固定した。Type 1と2の試料において,SFCA,SFCA-Iのプロファイル関数の半値全幅パラメーター(U, V, W)は,α-Fe2O3の値に統一した。XRD-Rietveld法は文献10)に詳しい。
2・3 Taylor-Matulis補正法Taylor and Matulis20)によると,i番目の結晶相の質量分率に対するXRDパターンのMA効果補正係数τiは,式(1)で表現される。
| τi=1Ai∫0Aie−(μi−μ¯)xdAi | (1) |
ここで,Aiは結晶相iの粒子体積,μiはi番目の結晶相の線吸収係数,μは試料の平均線吸収係数である。なおμは,TM補正法による補正前の質量分率から計算した。ここで,粒子が完全な球であると仮定すると,式(1)は,式(2)の様に計算できる。
ここで,X=(μi-μ)Riである。Riはi番目の結晶相の平均粒径である。補正係数τiを用いて,i番目の結晶相の質量分率は,式(3)のように表現できる。
| wi=siZiMiViτi∑j=1nsjZjMjVjτj=wi'τi∑j=1nwj'τj | (3) |
ここで,si,Zi,Mi,Vi,wi,wi'は,それぞれ結晶相iのXRD-Rietveld法におけるスケール因子,単位セルあたりの原子数,式量,単位セル体積,結晶相の補正後,補正前の質量分率,nは試料中の結晶相の数である。
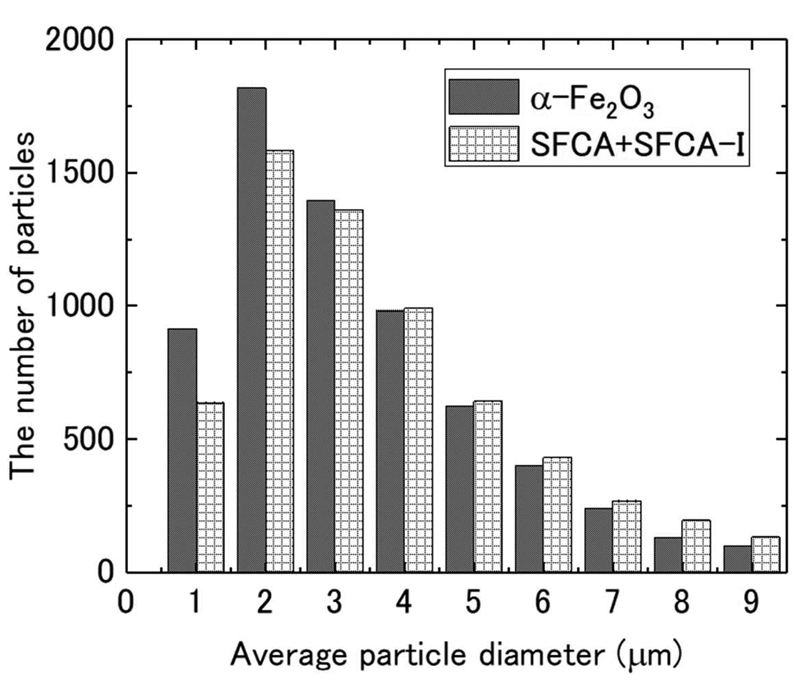
2・4 SEM-EDSによる粒径測定SEM-EDSを用いて,Table 2に示す試料No. 1中のα-Fe2O3と多成分カルシウムフェライト(SFCA+SFCA-I)の平均粒径を決定した。SEM-EDS測定には,Metal Quality Analyzer(MQA,Thermo Fisher Scientific)を用いた。試料は以下のように調整した。水中に分散させた試料No.1の粉末を,スポイトで採取し,カーボンテープを貼った10 × 10 × 1t mm3のSi基板上に滴下した。乾燥後,MQAの自動粒子機能を用いて,約7000粒子の粒径分布と組成を測定した。本研究では,EDSでFeとOのみが検出された粒子をα-Fe2O3とし,Al,Ca,Fe,OおよびSiが検出された粒子を多成分カルシウムフェライト(SFCA+SFCA-I)とした。異なる固溶状態を有する実機の焼結鉱では,組成のみでSFCAとSFCA-Iを識別できない場合が多いため,本研究では,SFCAとSFCA-Iをあえて区別出ずに補正を行うこととした。Fig.2にα-Fe2O3と多成分カルシウムフェライト(SFCA+SFCA-I)の粒度分布を示す。なお,α-Fe2O3と多成分カルシウムフェライトの平均粒径は,各粒子の面重心を通り直交する2つの方向と,それらと45°をなす2方向で測定した長さの平均値として定義した。
3. 結果と考察
3・1 マイクロアブソープション効果に対するTaylor-Matulis補正法の効果Fig.3に,Cu Kα X線源で測定した平均粒径の組み合わせの異なるα-Fe2O3とZnOの混合試料(質量比=1:1)のXRDパターンを示す。青線は,(a)平均粒径がそれぞれ0.5 μmと0.6 μmのα-Fe2O3とZnOの混合物のパターン,赤い線は,(b)平均粒径がそれぞれ20 μmと0.6 μmのα-Fe2O3とZnOの混合物のパターンである。(b)の場合,XRD-Rietveld法による質量分率はそれぞれFe2O3:35 mass%,ZnO:65 mass%と算出され,仕込みの質量分率から大きく外れた値となった。なお,XRD-Rietveld法のフィッティングの目安を示すRwp(重み付き残差)/S(Goodness-of-fit)は,それぞれ3.42%/2.52と2.56%/1.91であった。補正係数τは,Cu Kα1のX線エネルギー(8.048 keV)での線吸収係数とα-Fe2O3とZnOの平均粒径を考慮してTM補正法を行った。α-Fe2O3とZnOの線吸収係数と平均粒径およびτFe2O3とτZnOをTable 3に示す。なお,Type 1の実験の目的が,TM補正法の有用性の検証であるため,τFe2O3とτZnOは,各試料の仕込みの質量分率を用いて計算した。
Table 3. MA correction parameters for α-Fe
2O
3 and ZnO mixture specimens.
| Species | Linear absorption coefficient μ (cm-1) | Averaged particle diameter R (μm) | μ (cm−1) | Correction facto τ |
|---|
| α-Fe2O3 | 1119 | 0.5 | 689.5 | 0.984 |
| 1119 | 20 | 689.5 | 0.533 |
| ZnO | 260 | 0.6 | 689.5 | 1.02 |
Fig.4に,(a)α-Fe2O3とZnOの平均粒径がそれぞれ0.5 μmと0.6 μmである試料のXRDパターンにおけるTM補正法の適用前後の質量分率をそれぞれAとA’として示す。適用前後で質量分率は,ほぼ同じであった。Fe2O3とZnOで線吸収係数の差は大きかった一方で,それぞれの平均粒径がどちらも十分小さかったことが原因であると考えられる。(b)Fe2O3とZnOの平均粒径がそれぞれ20 μmと0.6 μmの試料のXRDパターンの場合のTM補正法適用前後の質量分率を,Fig.4にそれぞれBとB’として示す。TM補正法を適用しない場合は仕込みの質量分率を再現できなかった。一方,補正係τFe2O3とτZnOを式(3)に適用することで,平均粒径の差が大きかったとしても,仕込みの質量分率とほぼ同一の値を得た。粒径の効果は,μtp=1として定義される侵入深さtpにより理解できる。Cu Kαの場合,α-Fe2O3とZnOでそれぞれtp=18 μmとtp=70 μmである。つまり,X線を吸収する結晶相の平均粒径が侵入深さtpに較べて充分小さい場合には補正は不要であるが,粒径がtpと同程度に大きい場合には,MA効果を考慮することが特に重要であることがわかった。一方,Co Kαの場合,それぞれtp=89 μmとtp=46 μmであるので,補正が不要なα-Fe2O3の結晶粒径の範囲が大きい方に広がることが期待できる。
以上の議論から,平均粒径が(a)0.5 μmと0.6 μmおよび(b)20 μmと0.6 μmのα-Fe2O3とZnOの混合試料の定量結果の比較により,XRD-Rietveld法による定量値のTM補正法による補正が有効であることを確認した。
3・2 模擬焼結鉱試料におけるX線源の種類によるマイクロアブソープション効果の影響とTaylor-Matulis補正法の効果次に,Type 2の試料の実験結果を述べる。Table 4に,α-Fe2O3,SFCA,SFCA-Iの2・4節で述べた方法で測定した平均粒径,Cu Kα1およびCo Kα1の線吸収係数,6種の模擬試料に対して計算したTM補正法のτ係数をそれぞれ示す。α-Fe2O3と多成分カルシウムフェライトの平均粒径はそれぞれ4.1 μmと4.6 μmで,Cu Kαのデータについては約5~8%の補正が必要であることがわかった。それぞれの平均粒径は侵入深さtpの値よりも小さく,MA効果はType 1の試料ほど大きくはないと推測される。
Table 4. Correction factors,
τ, for
α-Fe
2O
3 and silico-ferrite of calcium and aluminum (SFCA+SFCA-I).
| species |
μ
(Cu Kα1)
(cm−1) |
μ
(Co Kα1)
(cm−1) | Averaged particle diameter R (μm) | No. |
μ(cm−1) | Correction factor τ for Cu Kα1 | Correction factor τ for Co Kα1 |
|---|
| α-Fe2O3 | 1119 | 225 | 4.1 | 1 | 945.7 | 0.948 | 1.006 |
| 2 | 961.2 | 0.953 | 1.007 |
| 3 | 929.1 | 0.943 | 1.006 |
| 4 | 934.9 | 0.945 | 1.006 |
| 5 | 926.6 | 0.943 | 1.006 |
| 6 | 919.6 | 0.941 | 1.006 |
| SFCA | 708 | 266 | 4.6 | 1 | 945.7 | 1.086 | 0.993 |
| 2 | 961.2 | 1.092 | 0.993 |
| 3 | 929.1 | 1.079 | 0.993 |
| 4 | 934.9 | 1.082 | 0.993 |
| 5 | 926.6 | 1.079 | 0.993 |
| 6 | 919.6 | 1.076 | 0.993 |
| SFCA-I | 837 | 268 | 4.6 | 1 | 945.7 | 1.038 | 0.992 |
| 2 | 961.2 | 1.044 | 0.993 |
| 3 | 929.1 | 1.032 | 0.992 |
| 4 | 934.9 | 1.034 | 0.992 |
| 5 | 926.6 | 1.031 | 0.992 |
| 6 | 919.6 | 1.029 | 0.992 |
以下の解析では,試料中の各結晶相の平均粒径は,6種とも同じ方法で粉砕および混合されたため,ほぼ同一であると見なすことができる。そこで,他の試料(No.2~6)についても試料No.1の平均粒子径を用いてTM補正を行った。SFCAおよびSFCA-Iの線吸収係数は,文献1,2)で報告されている組成と密度を基に計算した。
Fig.5に,α-Fe2O3,SFCA,SFCA-I1,2,26)からなる模擬焼結鉱試料(No.1~6)のCu Kα線で測定したXRDパターンを示す。Fig.5の下部には,先行研究で報告されているα-Fe2O3,SFCA,SFCA-IのXRDパターンの各ピーク位置(上からα-Fe2O3,SFCA,SFCA-I)を示している1,2,26)。Fig.6に,Cu Kαを用いて測定したモデル試料(No. 1)のXRDパターン(赤)とXRD-Rietveld法で再現したパターン(青),それらの残差パターン(ピンク)を示す。Fig.7とFig.8は,Fig.5とFig.6に対応したCo Kα源の結果である。Fig.5とFig.7のXRDパターンでは,No. 1-6においてSFCAとSFCA-Iの質量分率を反映してそれぞれ2θ=34.5, 40.5°付近のピークの強度が変化していることが確認できた。また,Fig.6とFig.8ではそれぞれのXRDパターンがXRD-Rietveld法で正しく再現できることが確認できた。Table 5に,Fig.5とFig.7に示すXRDパターンのXRD-Rietveld法におけるRwpとS係数を示す。
Table 5.
Rwp and
S factors of the Rietveld refinement of XRD patterns shown in
Figs. 5 and 7.
| Cu Kα | Co Kα |
|---|
| No. | Rwp | S | Rwp | S |
|---|
| 1 | 1.94 | 1.46 | 1.47 | 1.60 |
| 2 | 2.03 | 1.55 | 1.34 | 1.47 |
| 3 | 2.13 | 1.61 | 1.51 | 1.63 |
| 4 | 2.03 | 1.55 | 1.55 | 1.68 |
| 5 | 2.25 | 1.70 | 1.68 | 1.82 |
| 6 | 2.26 | 1.70 | 1.65 | 1.79 |
Fig.9に,各試料の仕込みの質量分率(Table 2)と式(3)とTable 4に示す補正係数を用いたTM補正法の適用前後の質量分率の関係を示す。Fig.9(a)と(b)は,それぞれCu KαおよびCo Kαでの結果である。Table 6は,Fig.9(a)と(b)の各質量率を整理したものである。Fig.9(a)と(b)の点線は,質量分率の±3 mass%の偏差を示している。Cu Kα線によるMA効果の補正前の質量分率は,線吸収係数が1119 cm-1と他の二相(SFCA,SFCA-I)より大きいα-Fe2O3の質量分率が実際の質量分率よりも小さく定量されていることがわかる。それに伴い,線吸収係数がα-Fe2O3より相対的に小さい多成分カルシウムフェライト(SFCA+SFCA-I)の質量分率は,実際の質量分率よりも大きく算出されている。これらの質量分率に対して,TM補正法を適用することにより,Fig.9(a)中の白抜きのプロットの様に,広い範囲にわたって,実際の質量分率とほぼ同一の質量分率が得られた。

Table 6. Mass fractions of the crystalline phases before and after the correction in the XRD measurements of type 2 samples (
α-Fe
2O
3: H, SFCA: S0, SFCA-I: S1).
| | Cu Kα | Co Kα |
|---|
| Initially-charged
mass fraction | Before correction | After correction | Before correction | After correction |
|---|
| H | S0 | S1 | H | S0 | S1 | H | S0 | S1 | H | S0 | S1 | H | S0 | S1 |
|---|
| No.1 | 50.0 | 25.0 | 25.0 | 45.9 | 28.0 | 26.1 | 48.7 | 26.0 | 25.3 | 51.7 | 24.0 | 24.3 | 51.4 | 24.2 | 24.5 |
| No.2 | 49.8 | 12.6 | 37.6 | 45.2 | 16.5 | 38.4 | 47.7 | 15.2 | 37.0 | 51.5 | 13.5 | 35.0 | 51.2 | 13.6 | 35.2 |
| No.3 | 49.8 | 37.5 | 12.7 | 44.4 | 41.3 | 14.3 | 47.5 | 38.6 | 14.0 | 52.2 | 35.2 | 12.6 | 51.8 | 35.5 | 12.7 |
| No.4 | 50.0 | 33.4 | 16.6 | 46.9 | 35.1 | 18.0 | 49.9 | 32.6 | 17.5 | 53.5 | 30.3 | 16.2 | 53.0 | 30.6 | 16.4 |
| No.5 | 50.0 | 39.8 | 10.3 | 44.7 | 43.8 | 11.5 | 47.8 | 41.0 | 11.2 | 51.8 | 37.6 | 10.6 | 51.5 | 37.8 | 10.7 |
| No.6 | 49.9 | 45.1 | 5.0 | 46.6 | 48.2 | 5.2 | 49.8 | 45.1 | 5.1 | 53.3 | 41.8 | 4.8 | 53.0 | 42.1 | 4.9 |
一方で,Co Kαでのデータでは,TM補正前の質量分率でも,実際の質量分率と±3 mass%以内で一致した。さらに,TM補正法を適用した場合もその効果は小さかった。これは,すべての結晶相でCo Kαにおける線吸収係数が小さいためであると考えられる。以上の結果から,Cu Kα線も用いても,本研究で述べたTM補正法を使用することで,各結晶相の質量分率を±3 mass%の確度で定量できることを確認した。したがって,MA効果は確かに各結晶相の質量分率値に影響するが,試料を十分細かく(粒径が5 μm以下程度)粉砕することにより,焼結鉱に含まれる主要な結晶相(α-Fe2O3,SFCA,SFCA-I)については,それら由来のXRDパターンの形状を大きく変化させるほどは影響しないことがわかった。これは,Cu Kα線を用いた測定でFe含有結晶相の線吸収係数が大きく,侵入深さtpが小さい条件でも,試料がそれ以上に十分細かい多結晶体であれば,XRD測定においてMA効果の影響を受けることが少ないためである。
このような評価条件でのXRD-Rietveld法による定量確度は,製銑プロセスにおいて,焼結鉱中の結晶相の質量分率と焼結鉱の特性の関係を調査する評価方法としては十分である。ただし,実機の焼結鉱にこの手法の適用する際には,以下に述べる点を十分考慮する必要がある。実際の焼結鉱における多成分カルシウムフェライト(SFCA,SFCA-I)は,高温の融液から非平衡の冷却過程を経て生成するため結晶性が非常に低い。加えて,焼これらの化学組成には幅がある。そのため,多成分カルシウムフェライト由来のピークは非常にブロードなピークとなり,XRD-Rietveld法によるフィッティングの際,SFCAおよびSFCA-Iの結晶構造の精密化によるピークフィッティングが実際の焼結鉱中のSFCAおよびSFCA-Iの質量分率を反映して適切に計算されるかについて留意する必要がある。
以上の結果から,Type 2の試料では,MA効果の影響が大きく定量性が低いであろうCu Kα線源を用いた場合でも,TM補正法を用いることにより,XRD-Rietveld法による定量確度が向上することを確認した。
4. 結言
本研究では,製銑プロセスへのXRD-Rietveld法による結晶相の定量への適用を念頭に,(1)α-Fe2O3とZnOの混合試料(Type 1)による線吸収係数と平均粒径差の関係,(2)模擬焼結鉱試料(Type 2)による質量分率に及ぼすX線源(Cu Kα, Co Kα)の影響とそれに対するTM補正法の効果,の二点を明らかにした。
Fe K-端吸収端近いエネルギーを有するCu Kα線の場合,試料に含まれるFe含有量によって結晶相の線吸収係数が大きく異なり,XRD-Rietveld法により定量された質量分率が,実際の質量分率を反映しない場合があることを確認した。Type 1の試料では,α-Fe2O3とZnOの平均粒径が両者で十分に小さい場合には,仕込みの質量分率とほぼ同一の質量分率が得られた。一方,平均粒径差が大きい場合には,各結晶相の実際の質量分率とは異なる値となった。ただし,混合試料の質量分率は,各相の線吸収係数と平均粒径を考慮したTM補正法を適用することにより,実際に近い値に補正できることを確認した。
SEM-EDS測定で得た各相の平均粒径と組成から計算した線吸収係数を考慮したTM補正法を考案し,α-Fe2O3,SFCA,SFCA-Iを含む模擬焼結鉱試料(Type 2)での検証を行った。この方法により,酸化鉄を多く含む試料にCu Kα線を用いた場合でも,±3 mass%以内の確度で質量分率が得られることを確認した。また,Co Kα線を用いた場合,TM補正法を適用しなくても,各結晶相の質量分率を±3 mass%以内の確度で定量できることを確認した。これは,Co Kα線のエネルギーにおける鉄系酸化物および多成分カルシウムフェライトの線吸収係数の差が小さいためである。以上より,試料に大きく吸収されるX線源を使用する場合でも,本研究で用いた補正方法により,各結晶相の質量分率の定量確度を向上できることを確認した。この方法は,モデル試料だけでなく,実際の焼結鉱をはじめとした様々な工業材料への適用が期待される。3・2節では,十分試料粉末を細かくすれば,MA効果を軽減して定量できると述べた。それは,本論文で報告したのは模擬焼結鉱試料であり,粉砕しても非晶質化する相は無かったので,十分細かくすればよかったからである。一方,実焼結鉱等の工業材料の場合,三相以上の相が含まれることがほとんどで,それらはそれぞれ機械的強度が異なっており,各相均一に微細に粉砕するのは容易で無い。加えて,粉砕しすぎると非晶質化する相もあるため,粉砕のみの試料を用いた場合よりも,ある程度まで粉砕した試料のXRDパターンを本論文での提案方法にて定量化する方が,試料の実際の性状をより正確に捉えられる方法であると考えられる。
謝辞
日本製鉄(株)の高橋貴文博士,金橋康二博士,原恭輔博士,樋口謙一博士,高山透氏,相本道宏博士,齋藤公児博士,日鉄テクノロジー(株)の岡崎潤博士,前橋工科大学の佐川孝広准教授,名古屋工業大学の井田隆教授には,XRD-Rietveld法と焼結鉱についての有益な議論をいただきましたので感謝の意を表します。また,SEM-EDS測定を行う上での技術的な助言と支援を賜った日本製鉄(株)の板橋大輔博士に感謝致します。
文献
- 1) J.D.G. Hamilton, B.F. Hoskins, W.G. Mumme, W.E. Borbidge and M.A. Montague: Neues Jahrb. Miner. Abh., 161(1989), 1.
- 2) W.G. Mumme, J.M.F. Clout and R.W. Gable: Neues Jahrb. Miner. Abh., 173(1998), 93. https://doi.org/10.1127/njma/173/1998/93
- 3) Y.Hida, J.Okazaki, K.Itoh and M.Sasaki: Tetsu-to-Hagané, 73(1987), 1893 (in Japanese). https://doi.org/10.2355/tetsutohagane1955.73.15_1893
- 4) L.X. Yang and C.E. Loo: ISIJ Int., 37(1997), 449. https://doi.org/10.2355/isijinternational.37.449
- 5) F.Zhang, S.An, G.Luo and Y.Wang: J. Iron Steel Res. Int., 19(2012), No. 4, 1.
- 6) F.Shen, G.Li, Z.Ding and L.Mu: J. Iron Steel Res. Int., 16(2009), No. 3, 1. https://doi.org/10.1016/S1006-706X(09)60035-2
- 7) I. Tonžetić and A. Dippenaar: Miner. Eng., 24(2011), 1258. https://doi.org/10.1016/j.mineng.2011.04.012
- 8) H.M. Rietveld: J. Appl. Crystallogr., 2(1969), 65. https://doi.org/10.1107/S0021889869006558
- 9) R.J. Hill and C.J. Howard: J. Appl. Crystallogr., 20(1987), 467. https://doi.org/10.1107/S0021889887086199
- 10) F. Izumi and K. Momma: Solid State Phenom., 130(2007), 15. https://doi.org/10.4028/www.scientific.net/SSP.130.15
- 11) N. Doebelin and R. Kleeberg: J. Appl. Crystallogr., 48(2015), 1573. https://doi.org/10.1107/S1600576715014685
- 12) L. Lutterotti, S. Matthies, H.-R. Wenk, A.S. Schultz and J. Richardson, Jr.: J. Appl. Phys., 81(1997), 594. https://doi.org/10.1063/1.364220
- 13) T.Degen, M.Sadki, E.Bron, U.König and G.Nénert: Powder Diffr., 29(2014), Suppl. S2, S13.
- 14) J.C. Taylor: Powder Diffr., 6(1991), 2. https://doi.org/10.1017/S0885715600016778
- 15) S. Ichikawa, D. Fujimura, A. Ohbuchi and T. Nakamura: ISIJ Int., 56(2016), 2228. https://doi.org/10.2355/isijinternational.ISIJINT-2016-392
- 16) T. Takayama, R. Murao and M. Kimura: ISIJ Int., 58(2018), 1069. https://doi.org/10.2355/isijinternational.ISIJINT-2017-717
- 17) L. Alexander and H.P. Klug: Anal. Chem., 20(1948), 886. https://doi.org/10.1021/ac60022a002
- 18) J.Als-Nielsen and D.McMorrow: Elements of Modern X-ray Physics, 2nd ed., John Wiley and Sons, Hoboken, (2011), 18.
- 19) G.W. Brindley: Lond. Edinb. Dublin Philos. Mag. J. Sci., 36(1945), 347. https://doi.org/10.1080/14786444508520918
- 20) J.C. Taylor and C.E. Matulis: J. Appl. Crystallogr., 24(1991), 14. https://doi.org/10.1107/S002188989000841X
- 21) N.V.Y. Scarlett and I.C. Madsen: Powder Diffr., 33(2018), 26. https://doi.org/10.1017/S0885715618000052
- 22) R.S. Winburn, D.G. Grier, G.J. McCarthy and R.B. Peterson: Powder Diffr., 15(2000), 63. http://doi.org/10.1017/S0885715600011015
- 23) R.S. Winburn, S.L. Lerach, B.R. Jarabek, M. Wisdom, D.G. Grier and G.J. McCarthy: Adv. X-ray Anal., 42(1998), 387.
- 24) I.C. Madsen, N.V.Y. Scarlett, L.M.D. Cranswick and T. Lwin: J. Appl. Crystallogr., 34(2001), 409. https://doi.org/10.1107/S0021889801007476
- 25) B.M. Pederson, K.J. Schaible and R.S. Winburn: Adv. X-ray Anal., 47(2003), 200.
- 26) D. Nie, T. Xue, Y. Zhang and X. Li: Sci. China, Ser. B: Chem., 51(2008), 823. https://doi:10.1007/s11426-008-0061-0
- 27) J. Albertsson, S.C. Abrahams and Å. Kvick: Acta Crystallogr. B, 45(1989), 34. https://doi.org/10.1107/S0108768188010109