Application of genome editing technology in human gene therapy
Article ID: 2020-007

Details
Article ID: 2020-007
In recent years, gene therapy drugs have finally been approved in Europe, the U.S., and Japan. In parallel with this, advances in genome editing technologies have enabled therapeutic strategies by gene knockout and gene repair, which were difficult with conventional so-called gene addition therapy. Worldwide, over 30 clinical trials of genome editing therapy have already been conducted, and some protocols have shown not only safety but also therapeutic efficacy. In the clinical application of genome editing, in addition to the technical hurdles of conventional gene therapy, there are problems specific to genome editing technology, such as the risk of introducing DNA mutations due to off-target activity of enzymes and the immune response to the artificial nucleases. It is necessary to consider the objective risk and benefit in comparison to existing therapeutic protocols. It is also essential to further develop technologies for therapeutic application in a wider range of diseases.
In recent years, genome editing has quickly become a popular biological tool. The applications range from basic science to therapeutic drugs, in medicine and other biological fields. The therapeutic application of genome editing can be considered a form of gene therapy, which has also been drawing increased attention. However, genome editing, like conventional gene addition therapy, has many technical challenges to overcome, from off-target activities to immunogenicity, before its potential can be fully realized. This minireview summarizes recent progress in genome editing therapy and discusses these challenges.
From Glybera (adeno-associated viral vector (AAV) for the treatment of familial lipoprotein lipase deficiency) in 2012 to Zynteglo (a lentiviral vector for the treatment of β-thalassemia) in 2019, gene therapy drugs are being approved one after another in Europe, the United States, and Japan. The recent success is based on the steady progress in basic research on gene therapy-related area over 20–30 years. In addition to conventional gene therapy using the overexpression of therapeutic genes, the development of therapeutic strategies utilizing genome editing technology is also rapidly advancing [1]. Gene therapy utilizing gene knockout or gene knockin is currently being evaluated in clinical trials, as described below. New genome editing tools, such as a base editor that enables single base substitution without introducing double-strand breaks into chromosomal DNA and a prime editor that enables the deletion and insertion of tens of bases [2], will also be used clinically in the near future. This paper introduces the recent preclinical and clinical studies of genome editing and discusses the problems that may arise in the clinical setting. There will also be issues on intellectual property related to artificial nucleases and vectors and problems related to vector production. However, I will leave these to other articles.
Among gene editing strategies, gene knockout by inaccurate non-homologous end-joining (NHEJ) has opened up new strategies for gene therapy. In addition, if homology-directed repair (HDR) using genome editing is used, the treatment of dominant genetic diseases and a safe and stable gene expression—due to site-specific chromosomal integration of the therapeutic gene—can be obtained (Fig. 1). As points to be considered in the clinical application of genome editing, there are potential problems unique to the genome editing technology, in addition to the problems of conventional gene therapy. Progress in the efficiency and safety of gene delivery methods (vectors) and advances in understanding on host immune responses to vectors and therapeutic genes are the keys to the recent success of gene therapy. In particular, it has been critical that the improvement of vector technology has made it possible to introduce genes into various target tissues with efficiencies close to 100%. Using this vector technology, artificial nucleases can also be introduced and expressed in target cells with high efficiency. However, successful genomic editing requires subsequent steps of efficient chromosome breaks and DNA repair (Fig. 1). Thus, in order for genome editing to be applied to a wider range of diseases, it is necessary to increase the rate of each of these steps.
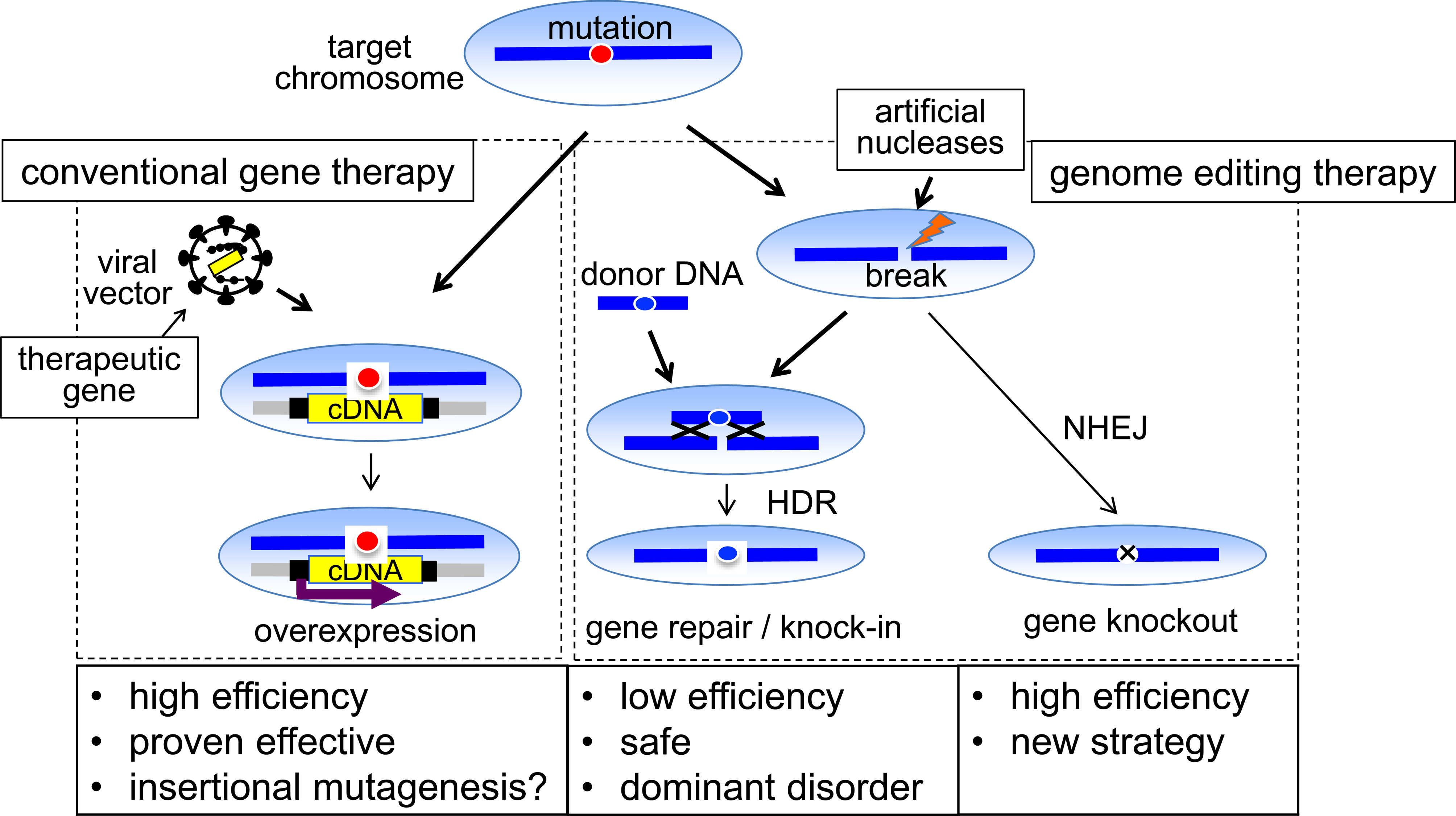
Comparison of conventional gene addition therapy and genome editing therapy for inherited diseases. Among the gene therapy strategies, genome editing technology added new options, such as efficient gene knockout and precise and seamless repair of DNA mutations, in gene therapy strategies.
The risks and benefits when applying genome editing to therapeutic treatment vary greatly depending on the target disease. In particular, when applied to advanced cancer treatment, the risk of the off-target mutations described below does not pose a problem. On the other hand, when treating genetic diseases in children, long-term therapeutic effects and safety must be considered. The content discussed in this paper is primarily intended for applications in genetic diseases, which have higher hurdles.
There have been numerous preclinical studies on genome editing therapy. The genome editing efficiencies reported in these studies are summarized in Fig. 2. For example, regarding ex vivo genome editing (a method in which cells taken out of the body are subjected to genome editing and then returned to the body) in human CD34-positive hematopoietic progenitor cells, primary/secondary transplantations into NOG/NSG -immunodeficient mice are often used to evaluate primitive progenitors. A population of human-derived cells that are later detected in the mouse blood are regarded as progenitors that are closer to the real hematopoietic stem cells. Currently, gene knockout with >90% efficiency and 10–20% HDR is possible in this type of cells. Because X-linked severe combined immunodeficiency can be potentially cured when 10% of the stem cells in the patient’s bone marrow are functionally normal, the current level of gene repair efficiency might be enough to expect therapeutic effects [3]. In the liver, the HDR efficiency is ~10% for in vivo genome editing (direct genome editing method in a body) in mice. In another example, an artificial nuclease called a meganuclease was used to knockout the PCSK9 gene with approximately 40% efficiency in the monkey liver [4]. In the muscle, cases of successful treatment in large animals, such as the excision of mutant exon in a dog model of muscular dystrophy, have been reported. The efficiency at the DNA level is still low, at approximately 10%. However, since muscle fibers are multinucleated cells, therapeutic effects are likely to appear [5]. In addition, reports on in vivo base editing (e.g., in post-mitotic cochlea in mice [6]) are emerging. Thus, at the animal level, the effectiveness of various types of genome editing therapy has been demonstrated.
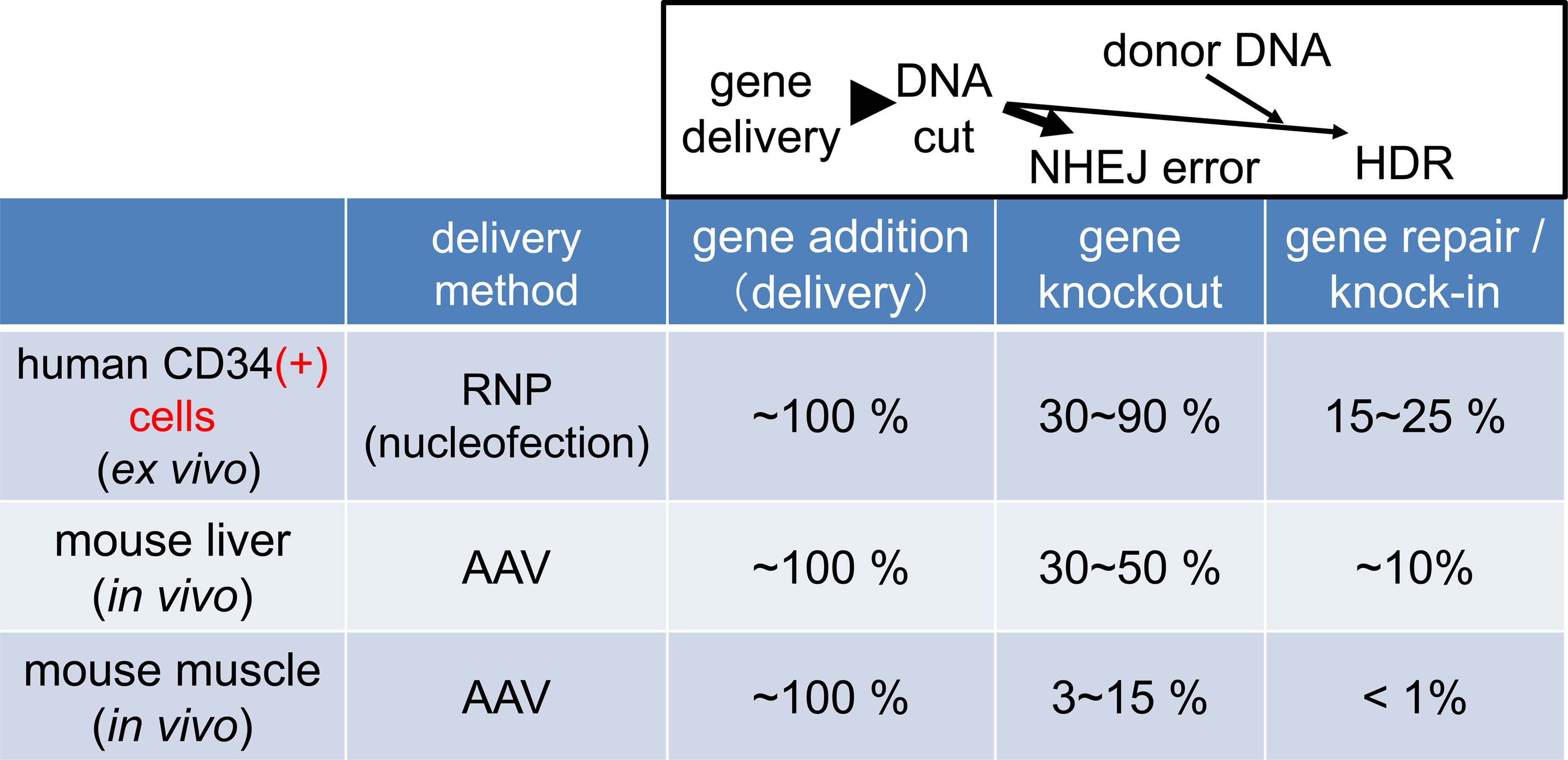
Recently reported efficiency in preclinical studies of genome editing in ex vivo hematopoietic progenitors and in vivo in the muscle and liver. The efficiency in these studies is still much lower in comparison to gene transfer efficiency because additional steps are taken.
For humans, clinical studies are being carried out on protocols that are likely to have therapeutic effects, even with low genome editing efficiencies. Representative examples of ongoing applications include the treatment of HIV-infection and the establishment of universal chimeric antigen receptor-T cells (CAR-T). In the former, immune cells are made resistant to HIV infection by knocking out the CCR5 co-receptor gene essential for HIV infection using zinc-finger nuclease (ZFN) or CRISPR (NCT01044654, NCT00842634, NCT02225665, NCT02500849, NCT03164135, etc.). Although it has been regarded as a highly promising treatment, the results reported in the literature thus far, show that the therapeutic effects have not been so remarkable [7, 8]. The universal CAR-T is a CAR-T cell line that can be commonly used in various patients without being restricted by human leukocyte-type antigens, unlike the conventional method that requires the establishment of CAR-T for each patient. In the universal CAR-T cells, which were already published, the T cell receptor α chain gene of CD19-targeted CAR-T cells was knocked out by TALENs. These universal CAR-T cells have been used in pediatric patients with B-cell acute lymphocytic leukemia and have produced significant therapeutic effects [9]. In addition to the above examples, cancer immunotherapy using T cells, gene knocked out by CRISPR, was recently reported (NCT03399448) [10]. In this trial, an NY-ESO-1 targeting T cell receptor gene was isolated from cancer-specific T cells and introduced into patient-derived T cells with a lentiviral vector. At the same time, two T receptor genes (α, β) and the PD-1 gene were knocked out to promote a therapeutic effect. In the first 3 patients (advanced multiple myeloma, n=2; myxoid round cell liposarcoma, n=1), the knockout efficiency was 15–45% for each locus, and the transduction efficiencies of NY- ESO-1 T cell receptors was 1.4–4.5% [10]. Although no clear treatment effect was obtained for these terminally ill cancer patients, there were no noticeable side effects.
Regarding hematopoietic genetic diseases, clinical trials of gene therapy using genome editing have begun for sickle cell disease and β-thalassemia, which are caused by an abnormal β-globin gene. The strategies for both diseases are the same, knocking out the erythroid-specific enhancer sequence of the BCL11A gene, which suppresses the expression of γ-globin that constitutes fetal hemoglobin in adult cells. As a result, instead of adult type hemoglobin with quantitative or qualitative abnormalities in these diseases, fetal hemoglobin is produced to cure the disease [11]. Both ZFN (Sangamo Therapeutics, NCT03432364 and NCT03653247) and CRISPR/Cas9 (CRISPR Therapeutics, NCT03655678 and NCT03745287) are utilized in each of the clinical trials. Especially for sickle cell disease, if the efficiency of HDR becomes higher, the ultimate gene repair therapy that accurately repairs the mutant gene sequence will become possible.
The first example of in vivo genome editing therapy, which has high technical hurdles, has also been started. This is a protocol, in which a therapeutic gene is knocked in at the Albumin locus of the liver of a patient with mucopolysaccharidosis, an inherited metabolic disease, using ZFNs (NCT02702115). Low HDR efficiencies in the liver, perhaps a few percent, are compensated for by high gene activity at the knockin target Albumin locus. This strategy can be a platform that enables various diseases to be similarly treated, using the same ZFNs and homology arms on the donor DNA. This trial has been drawing much attention because it is the first in vivo clinical application and protocol using HDR. Recently, it has been reported that treatment to knock out the causative CEP290 gene mutation has been started for Leber congenital amaurosis 10 (LCA10), which is caused by a dominant mutation in the CEP290 gene (NCT03872479, see reference [12] for treatment strategy). In this example, the AAV5 vector encoding SaCas9 and gRNA to delete that mutated exon was injected subretinally. This is the first in vivo application of CRISPR. As in the case of hemoglobinopathies, a gene knockout strategy, which has lower hurdles in terms of efficiency and safety than gene repair, is used. According to the American Society of Gene & Cell Therapy (ASGCT) Policy Summit last November, more than 30 protocols for genome editing clinical trials are currently in progress (type of nuclease: CRISPR, n=19; ZFNs, n=9; TALENs, n=5; and other, n=2; target disease: abnormal hemoglobinosis, n=5; cancer, n=16; infectious disease, n=7; ophthalmic disease, n=1; and other, n=8). This number is expected to increase steadily. Many of these protocols, like conventional gene therapy, use viral vectors to achieve high delivery efficiency and high nuclease expression levels. However, a stable gene expression, which is advantageous in conventional gene therapy, may be disadvantageous in the case of the expression of nuclease because of immunogenicity and off-target effects. There will be a high demand for the development of technologies capable of transiently and strongly expressing a nuclease, which has received relatively little attention in the development of gene therapy vectors.
Establishing a protocol to obtain a therapeutic effect is the highest priority when developing a therapeutic strategy. However, since it is used in humans, “safety” is the most important priority. Problems in gene therapy as a whole, include immunogenicity and cytotoxicity derived from the vector. In addition, when chromosomally integrating vectors, such as retrovirus or lentivirus vectors are used, genotoxicity (the adverse effect on chromosomal DNA) becomes a problem. Additional problems associated with the genome editing technique include immunogenicity, cytotoxicity, and genotoxicity (so-called off-target mutation) of artificial nucleases. Furthermore, when donor DNA for HDR is used, genotoxicity due to the integration of the donor DNA into the chromosome becomes an issue.
Off-target mutations by nucleases can be analyzed in various ways [13]. The most accurate method, in theory, would be the analysis of the whole-genome sequence of gene-edited cells. However, even if one mutation is found with ×100 coverage of the whole genome, the detection sensitivity will be at most 1%. Considering that the number of target cells exceeds the order of 108 cells in many clinical applications, at this sensitivity, mutations cannot be detected in nearly 106 cells. Considering the possibility that only one mutant cell eventually causes cancer, this detection sensitivity is insufficient. On the other hand, regarding the prediction of the off-target sites, a method of identifying genome sequences similar to the target sequence by a computer analysis is convenient. However, it is recognized that many of the sites predicted in silico do not match the actual off-target site [14]. As a comprehensive and unbiased off-target prediction, methods have been developed to express the artificial nuclease in cultured cells to detect the cleavage site (BLESS [15], GUIDE-seq [14], etc.), or cut the DNA extracted from the target cells with the artificial nuclease in a test tube (Digenome-seq [16], CIRCLE-seq [17], etc.). In particular, the latter can detect differences due to SNP in the genomic sequence between individuals. These methods are only applicable in pre-screening of off-target sites. After using these methods, nucleases or guide RNAs that have the smallest number of off-target sites, especially those that are not located near cancer-related genes, will be chosen for further use. In order to examine the frequency of off-target mutations in cells after the actual genome editing process, deep sequencing is performed for each predicted site. However, the detection sensitivity is approximately 0.1%, based on the error rate of next-generation sequencing. As with the above-mentioned example, when genome editing is performed on 108 cells, mutations with a frequency of <105 cannot be detected at this error rate. In addition, it is difficult to analyze off-target sites when using a genome editing tool such as base editing, which does not introduce double-strand breaks into chromosomal DNA. Recently, the relationship between the p53 gene and genome editing has been reported. Cells with a normal p53 gene are likely to die due to DNA double-strand breaks, suggesting that cells with successful genome editing are likely to have an abnormality in the p53 pathway [18]. However, at least in human hematopoietic stem progenitor cells, this problem can be avoided by selecting targets with low off-target mutations [19]. In addition, DNA double-strand break repair errors were reported to result in deletions of several kilobase pairs or larger in size [20], suggesting that frequent chromosomal translocations may occur. The most problematic result of off-target mutations may be oncogenesis, but the above methods only analyze mutations at the DNA level. It is difficult to distinguish oncogenic and non-oncogenic mutations.
Off-target mutations by the nucleases are not the only sources of DNA mutations. When using donor DNA (double-stranded DNA or AAV vector), it is known that they are incorporated into on-target and off-target sites as frequently as 50% (0.5 sites per cell) of some cells (Fig. 3) [21]. In addition, mutations that occur during DNA replication during cell division. Assuming that replication errors occur at a frequency of 1 per 1010 bases per DNA replication [22], it is calculated that there are approximately 20 random mutations per cell after about 30 cell divisions (corresponding to expansion from 1 to 109 cells) (Fig. 3). This is orders of magnitude higher than the off-target mutation frequencies of artificial nucleases, which are considered to be less than 0.1% per cell. Therefore, stem cell therapy using iPS cells, after the process of iPSC induction, has mutation rates at an order of magnitude higher than in vivo genome editing. For this reason, cell cultures should be minimized, even in ex vivo genome editing. Finally, there are approximately 0.1% DNA sequence variants on genomic DNA among individuals (106 locations per cell) [22], making it difficult to find and interpret off-target mutations (Fig. 3). To our knowledge, there have been no reports on cellular transformation/tumorigenesis caused by off-target mutations in preclinical or clinical genome editing therapy. For this reason, it is recently considered that a carefully designed artificial nuclease would be associated with a very low risk of carcinogenesis due to off-target mutations, when the above-described comprehensive and unbiased screening methods are used.
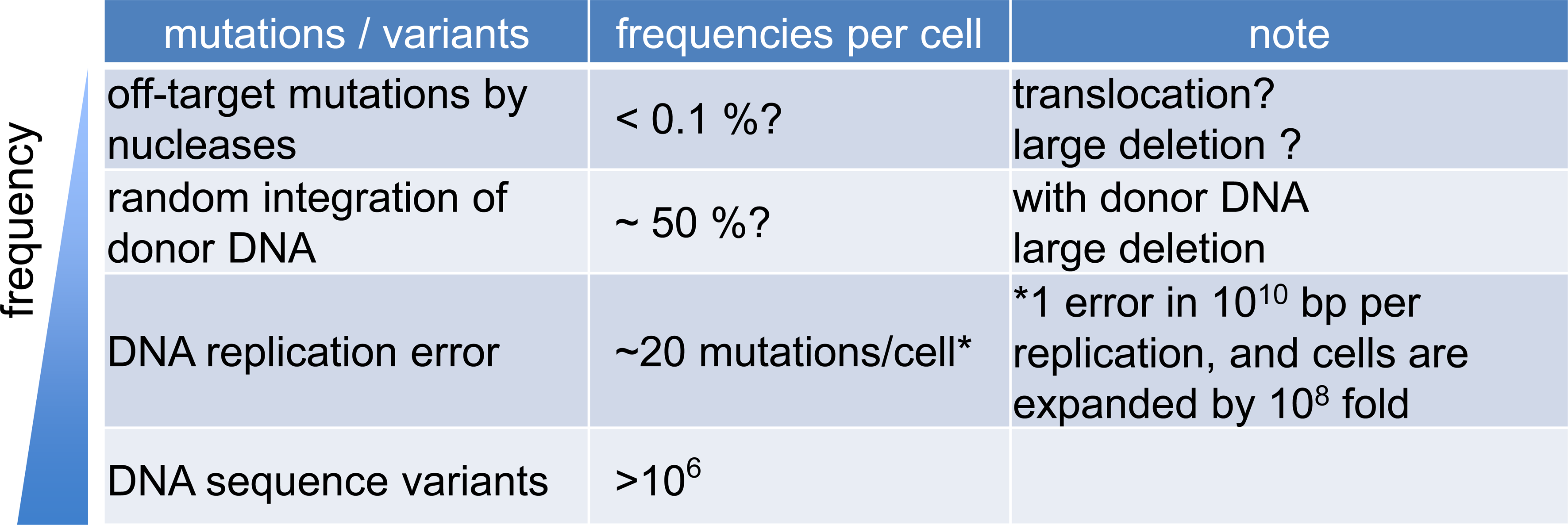
The frequency of mutations and variants, which potentially arise after therapeutic genome editing. In general, the frequency of off-target mutations by nucleases is not much higher than other types of mutations/variants.
The safety of the ex vivo method may be similar to the case of using ES cells or iPS cells for therapeutic purposes. In 2013, the Cell Tissue Processing Product Special Committee of the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan issued a report “Current Perspective on Evaluation of Tumorigenicity of Cellular and Tissue-based Products Derived from Induced Pluripotent Stem Cells (iPSCs) and iPSCs as Their Starting Materials”. According to this report, the criteria to evaluate the safety of stem cell products are genomic instability and mutations in a panel of cancer-related genes [23]. To evaluate the safety of gene-edited hematopoietic progenitors, a group in the U.S. transplanted as many genome-edited cells as planned for transplantation into patients into 100 or more NSG mice, which were observed for 5 months [24]. As mentioned above, only off-target mutations associated with the transformation of genome-edited cells are problematic. Thus, it is more realistic and sensitive to transplant the edited cells into animals than to analyze the off-target candidate sites of unknown biological significance by deep sequencing. However, for in vivo genome editing (e.g., liver and muscle), it is almost impossible to transplant the patient-equivalent number of genome-edited cells into animals, as in the case of hematopoietic progenitors. Furthermore, as described above, most in vivo genome editing performed thus far involves the continuous expression of artificial nucleases using viral vectors, which might result in the accumulation of off-target mutations. The development of assay systems to assess the safety of in vivo genome editing is a major issue for the future.
Increasing the number of emerging high-fidelity nucleases might eventually solve the problem of off-target mutations. However, the main problem will be the problem of immune response. As mentioned above, one of the most important themes in gene therapy research has been the immune response against gene therapy products by the host. Immunogenicity is unavoidable when artificial nucleases are non-human proteins, especially those of bacterial origin. In a study that actually examined the prevalence of anti-Cas9 antibodies and anti-Cas9 T cells in normal individuals, more than half already possessed humoral and cellular immunity to Cas9, depending on the target population [25]. On the other hand, recent results of preclinical in vivo genome editing of muscle showed that an immune response was elicited in adult mice but not neonatal mice [26]. Looking back at the history of gene therapy research, even if successful treatment with inbred mouse strains is obtained, it is quite challenging to obtain similar results in large animals or humans, because they are often outbred and have a more complicated immune system. The difficulty in producing gene therapy drugs in high quantities is another challenge. The development of nucleases with lower immunogenicity and a method of efficiently introducing them as protein products rather than as a gene are desired to circumvent these issues [27]. In any case, it is difficult to completely prevent the immune response, and it is necessary to consider whether there are advantages in choosing a genome-editing treatment—even with a strong immunosuppressive drug—in comparison to other therapies.
Almost any therapy is accompanied by various degrees of risk. Thus, it is necessary to compare the risks and benefits between genome editing and existing treatment methods, including gene (addition) therapy. Of course, there is a large benefit in diseases that can be treated by genome editing alone, and dominant genetic diseases fall into this category. Immune diseases, such as CD40 ligand deficiency, which require a controlled gene expression, are also target diseases for which genome editing can have large benefits [28]. Considering the risk, ex vivo genome editing for hematological malignancies and infectious diseases have far lower hurdles in comparison to in vivo treatment of genetic diseases. In addition, disorders, in which normal cells (gene repair cells) have a growth advantage among mutant cells would be good candidates because of the relatively low efficiency of gene repair at the present time. In addition, disorders, such as hemophilia, in which therapeutic efficacy is expected, even with low gene expression levels, are considered appropriate targets. However, in many of these diseases, conventional gene addition therapy has already shown effective results. There is a need for research that shows that genome editing is safer and more effective than conventional therapy. Nonetheless, with further advancements in genome editing technologies, there is no doubt that genome editing will be increasingly applied in clinical areas.