Abstract
The development of oligonucleotide therapeutics (ONTs) has advanced recently. Various
ONTs (e.g., antisense oligonucleotides, small-interfering RNA, and microRNA) exert their
pharmacological effects via hybridization with mRNA sequences, and they can also bind to
unintended mRNA sequences owing to sequence homology. For this reason, the safety of ONTs
should be evaluated by judging hybridization-dependent on- and off-target toxicity in
preclinical studies. As the off-target toxicity is unique to ONTs, it is difficult to
assess their safety with the current guidelines established for small molecules and
biotechnology-derived pharmaceuticals; thus, several research groups, such as the
Oligonucleotide Safety Working Group in the Drug Information Association (DIA), have
proposed concepts for the preclinical safety evaluation of ONTs. Although there are
currently no specific International Council for Harmonisation of Technical Requirements
for Pharmaceuticals for Human Use (ICH) guidelines for ONTs, the ICH S6 guideline states
that “the principles outlined in this guidance may also be applicable to oligonucleotide
drugs.” Recently, a preclinical safety guideline for ONTs has been developed by a Japanese
working group to address the issues associated with the ICH S6. Here, the preclinical
safety assessments of mRNA-targeting ONTs are discussed based on this guidance.
Introduction
Oligonucleotide therapeutics (ONTs) are a groundbreaking therapy, for which more than 100
clinical trials are currently being conducted worldwide. To date, ten ONTs have been
approved for marketing by regulatory agencies, most of which are either antisense
oligonucleotides or small interfering RNA (siRNA) that target mRNA (Table 1). In Japan, Spinraza® (nusinersen), which
treats a rare disorder called spinal muscular atrophy, was the first antisense ONT approved
for marketing in July 2018. Last year 2019, Onpattro® (patisiran) was approved as
the first siRNA ONT, indicated for the treatment of polyneuropathy caused by hereditary
transthyretin-mediated amyloidosis. Although the knowledge required for ONT development is
gradually accumulating, there are still no standardized procedures for the preclinical
safety evaluation of ONTs. ONTs are highly target-specific, similar to biotechnology-derived
pharmaceuticals (biopharmaceuticals), but have the toxicological characteristics of new
chemical entities (NCEs) because most ONTs are composed of nucleotides with various chemical
modifications (e.g., 2’-methoxyethyl-modified nucleotides). Therefore, it is not appropriate
to automatically apply the conventional guidelines for biopharmaceuticals or small molecule
pharmaceuticals to evaluate the safety of ONTs. However, currently, the International
Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH)
S6 guideline states that “the principles outlined in this guidance may also be applicable to
oligonucleotide drugs”.
Table 1.
Summary of approved oligonucleotide therapeutics (as of April, 30, 2020)
| Generic name |
Trade name |
Type |
Target |
Indication |
First approved |
| Fomivirsen |
Vitravene |
Antisense |
CVM IE2 mRNA |
Cytomegalovirus retinitis in patient with
AIDS |
US: 1998 |
| EU: 1999 |
| Pegaptanib |
Macugen |
Aptamer |
VEGF protein |
Neovascular (wet) age-related macular
degeneration |
US: 2004 |
| EU: 2006 |
| JP: 2008 |
| Mipomersen sodium |
Kynamro |
Antisense |
ApoB-100 mRNA |
Homozygous familial hypercholesterolemia |
US: 2013 |
| Eteplirsen |
Exondys 51 |
Antisense |
Dystrophin pre-mRNA |
Duchenne muscular dystrophy |
US: 2016 |
| Nusinersen |
Spinraza |
Antisense |
SMN2 pre-mRNA |
Spinal muscular atrophy |
US: 2016 |
| EU: 2017 |
| JP: 2017 |
| Inotersen |
Tegsedi |
Antisense |
TTR mRNA |
Polyneuropathy of hereditary
transthyretin mediated amyloidosis |
US: 2018 |
| EU: 2018 |
| Patisiran |
Onpattro |
siRNA |
TTR mRNA |
Polyneuropathy of hereditary
transthyretin mediated amyloidosis |
US: 2018 |
| EU: 2018 |
| JP: 2019 |
| Volanesorsen |
Waylivra |
Antisense |
Apo-CIII mRNA |
Familial chylomicronemia syndrome |
EU: 2019 |
| Hepatitis B Vaccine (Recombinant), Adjuvanted |
HEPLISAV-B |
Vaccine |
Hepatitis B surface antigen |
Prevention of infection caused by all known subtypes
of hepatitis B virus |
US: 2019 |
| Golodirsen |
Vyondys 53 |
Antisense |
Dystrophin pre-mRNA |
Duchenne muscular dystrophy |
US: 2020 |
The preclinical safety assessment for ONTs has been discussed in Europe and the United
States [1,2,3,4,5,6,7,8]. In Japan, the Ministry of Health, Labor and Welfare recently issued a
preclinical safety guideline for ONTs [9]. This
guideline applies to ONTs that induce a biological reaction by hybridization with the target
sequence of DNA or RNA in the absence of novel protein synthesis. Specifically, antisense
oligonucleotides, siRNAs, and microRNAs are applicable. The principles outlined in this
guidance is also applicable to aptamer and decoy. This review will provide an overview of
the considerations for the preclinical safety assessment of the mRNA-targeting ONTs
described in this guideline.
Toxicity Classification of ONTs and Evaluation Strategy
To evaluate the preclinical safety of ONTs, it is necessary to understand the similarities
and differences between ONTs and other pharmaceuticals, such as NCEs and biopharmaceuticals.
Generally, biopharmaceuticals are highly specific to their target and exert mechanism-based
toxicity (known as on-target toxicity), whereas NCEs sometimes cause unexpected biological
functions (known as off-target toxicity), which are difficult to predict based on their
properties. As ONTs have the properties of both NCEs and biopharmaceuticals, it is necessary
to evaluate their preclinical safety regarding on- and off-target toxicity (Table 2).
Table 2.
Comparison of general characteristic of chemical entities, oligonucleotides and
biopharmaceuticals
|
Chemical entities |
Oligonucleotides |
Biopharmaceuticals |
| Modality |
Synthetic chemicals |
Synthetic chemicals |
Monoclonal antibodies, Fusion proteins etc. |
| Synthesis |
Chemical |
Chemical |
Biotechnological |
| Molecular weight |
<1,000 Da |
500–15,000 Da |
>1,000 Da |
| Target selectivity |
Low |
Low -High |
High |
| Species Specificity |
Non-specific |
Non-specific / Specific |
Specific |
| Typical animal |
Rodent and non-rodent |
Rodent and non-rodent |
Non-human primate |
| Metabolism |
Inactive and active metabolites |
Inactive and active metabolites |
Amino acids |
| Immunogenicity |
Rare |
Possible |
Possible |
| Toxicity |
Off-target |
Off-target/On-target |
On-target |
Various ONTs, such as antisense oligonucleotide, siRNA, aptamer, and decoy, hybridize
with specific sequences of mRNA and induce on-target toxicity owing to exaggerated
pharmacology. As the on-target toxicity of ONTs is similar to that of biopharmaceuticals,
the principles described in the ICH S6 (R1) guideline could be applied to evaluate the
on-target toxicity of ONTs [10]. For example, in
terms of species selection, it is appropriate to compare target sequences between animal
species and choose pharmacologically relevant animals for toxicity studies. When no
pharmacologically relevant animal species is available, a surrogate evaluation would be
recommended as described in the ICH S6 (R1) guideline. However, although a surrogate is
useful for hazard detection, it is not for quantitative risk assessment, as in the case of
biopharmaceuticals.
2) Hybridization-dependent off-target toxicity
Owing to the high homology between RNA sequences, ONTs can also bind to unintended mRNA
sequences and cause hybridization-dependent off-target toxicity. Although
hybridization-dependent off-target toxicity studies are crucial for human safety
evaluation, those using laboratory animals are of limited value because of genetic
differences between humans and animals. To identify hybridization-dependent off-target
toxicity, the Pharmaceuticals and Medical Devices Agency generally recommends in
silico screening with a representative public database (e.g., RefSeq and
GENCODE) and algorithm (e.g., GGGenome, and siDirect), then analyzing the results with an
in vitro array (e.g. quantitative-polymerase chain reaction (PCR),
microarray, reporter assay, next-gen transcriptome profiling methods such as RNAseq) using
human cells (Fig. 1). When candidates are
isolated, various data, such as information on the knock-out animal, human genetic
disease, target biology, and mechanism of action, should be utilized to evaluate the risk
to humans. If the weight of evidence approach suggests any clinical concern, an
appropriate risk management plan or optimized oligonucleotide sequence should be
considered to avoid the risk.
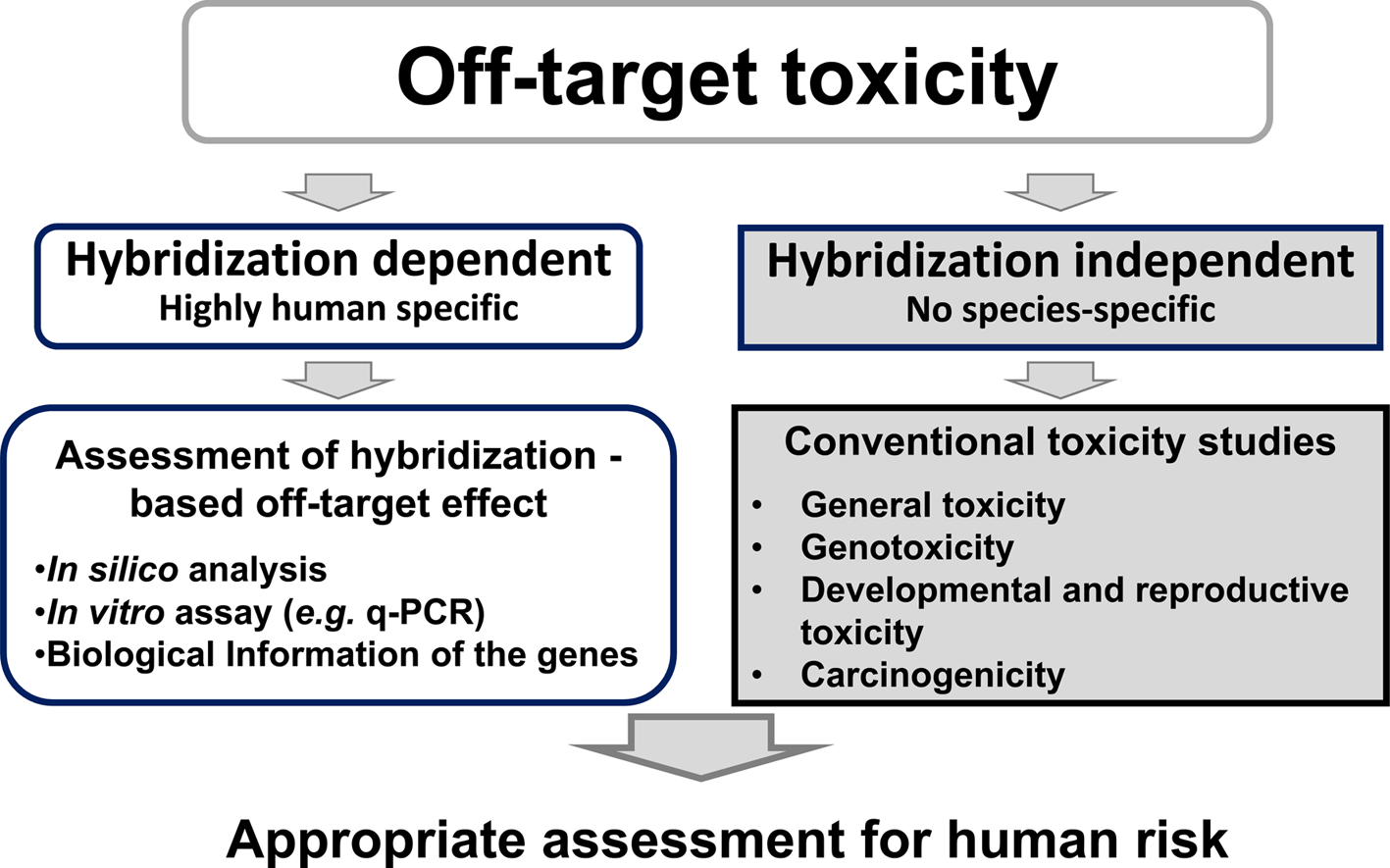
Off-target toxicity caused by any physicochemical property of ONTs, such as class effects
which caused by certain chemical structure (e.g., Toll-like receptor-mediated innate
immunity, prolonged activated partial thromboplastin time (aPPT), hepatotoxicity, and
nephrotoxicity) or toxicity from chemical modifications, can be detected with a
conventional toxicity study using laboratory animals (Fig. 1).
Study Design for Preclinical Safety Assessment of ONTs
To determine the animal species, dosing route, duration of treatment, and frequency of
administration for the preclinical safety evaluation, it is necessary to consider the
predicted human pharmacokinetic profile of the ONT [11,12,13]. The route and frequency of administration should be as close as possible to
those proposed for clinical use. When the pharmacokinetic profiles differ greatly between
humans and animals, it may be appropriate to consider other study designs to maintain the
level of exposure appropriate for the assessment of toxicity (e.g., frequent dosing).
1) High dose selection
To detect the off-target toxicity of chemically modified ONTs, it is necessary to follow
the ICH M3 guideline for NCEs [14]. In a
repeated-dose toxicity study, this guideline recommends a high dose that provides the
maximum tolerated dose, limiting dose (1,000 mg/kg/day), exposure saturation, and maximum
feasible dose or a dose that is 50-fold greater than the clinical exposure (see the ICH M3
guideline).
2) Species selection
In principle, two animal species should be used for the general toxicity testing of ONTs,
as in the case of NCEs and biopharmaceuticals. This strategy is intended to enhance the
prediction of human safety because chemically modified ONTs often show off-target toxicity
and their sensitivity and specificity are different among animal species. To evaluate
on-target toxicity, it is necessary to use the species in which the pharmacological effect
of the clinical candidate can be expressed. When at least one of the two species used for
the toxicity study is pharmacologically relevant for the clinical candidate, on-target
toxicity can be assessed. However, if no pharmacologically relevant species is available
for the clinical candidate, a safety assessment with surrogates should be considered.
3) Use of surrogates
ONTs are often species-specific because of their high target-specificity and preclinical
safety assessments using surrogates may be performed if relevant animal species with
pharmacological effects cannot be obtained with clinical candidates. However, as the
surrogate is never identical to the clinical candidate, it could induce surrogate-specific
off-target toxicity. Therefore, toxicological results using surrogates should be carefully
interpreted in the context of the overall pharmacology and safety assessment.
Safety Evaluation of Impurities
Most ONTs contain various impurities, such as product-related substances [e.g., n+1, n-1
mer impurities], small organic impurities, residual solvents, and elemental impurities. As
it is difficult to characterize each product-related substance, their safety should be
assessed in preclinical studies with ONTs. The safety of other impurities can be evaluated
with reference to ICH Q3A, ICH Q3B, and ICH M7 for small organic impurities, ICH Q3C for
residual solvents, and ICH Q3D for elemental impurities [15,16,17,18,19].
Conclusion
Based on the principles of the “case-by-case basis assessment” and “welfare of animals”,
the Japanese guideline provides points to consider to improve the preclinical safety
assessment of ONTs. However, continuous efforts are required to identify the limitations of
conventional toxicological methods and advance the proper safety evaluation of ONTs.
Conflict of Interest
The author declares no conflict of interest to this report. The views expressed herein are
the result of independent work and do not necessarily represent the views and findings of
the Pharmaceutical and Medical Devices Agency.
Acknowledgements
I am grateful to colleagues of the Japanese working group for ICH S6 and related issues:
Drs. Yoko Hirabayashi, Osamu Fueki, Misaki Naota, Kazuto Watanabe, Kiyoshi Kinoshita,
Mutsumi Suzuki, Takahiro Nakazawa, Satoshi Obika, Teruyo Arato, Aki Fujisaka, Kosuke Ito and
Hiroshi Onodera for valuable advice. I would like to thank Ms. Tamara Black for English
language editing. This work was supported by the Japan Agency for Medical Research and
Development (AMED, Promotion Project “Pharmaceutical Regulations Harmonization and
Evaluation Research Project”).
References
- 1. Schubert,
D., Levin,
A. A.,
Kornbrust,
D., Berman,
C. L.,
Cavagnaro,
J., Henry,
S., Seguin,
R.,
Ferrari, N.
and Shrewsbury, S.
B.
2012. The Oligonucleotide Safety Working Group
(OSWG). Nucleic Acid Ther.
22: 211–212.
- 2. Lindow,
M.,
Vornlocher, H.
P., Riley,
D.,
Kornbrust, D.
J., Burchard,
J.,
Whiteley, L.
O., Kamens,
J.,
Thompson, J.
D., Nochur,
S., Younis,
H., Bartz,
S., Parry,
J.,
Ferrari, N.,
Henry, S.
P. and Levin,
A. A.
2012. Assessing unintended hybridization-induced biological
effects of oligonucleotides. Nat.
Biotechnol.
30: 920–923.
- 3. Kornbrust,
D.,
Cavagnaro,
J., Levin,
A., Foy,
J., Pavco,
P.,
Gamba-Vitalo,
C. and
Guimond,
A.
2013. Oligo safety working group exaggerated pharmacology
subcommittee consensus document. Nucleic Acid
Ther.
23: 21–28.
- 4. Alton, E.
W., Boushey,
H. A.,
Garn, H.,
Green, F.
H., Hodges,
M., Martin,
R. J.,
Murdoch, R.
D., Renz,
H.,
Shrewsbury, S.
B., Seguin,
R.,
Johnson, G.,
Parry, J.
D., Tepper,
J., Renzi,
P.,
Cavagnaro,
J. and
Ferrari,
N.
2012. Clinical expert panel on monitoring potential lung
toxicity of inhaled oligonucleotides: consensus points and
recommendations. Nucleic Acid Ther.
22: 246–254.
- 5. Berman, C.
L., Cannon,
K., Cui,
Y.,
Kornbrust, D.
J., Lagrutta,
A., Sun,
S. Z.,
Tepper, J.,
Waldron, G.
and Younis, H.
S.
2014. Recommendations for safety pharmacology evaluations of
oligonucleotide-based therapeutics. Nucleic Acid
Ther.
24: 291–301.
- 6. Cavagnaro,
J., Berman,
C.,
Kornbrust,
D., White,
T.,
Campion, S.
and Henry,
S.
2014. Considerations for assessment of reproductive and
developmental toxicity of oligonucleotide-based therapeutics.
Nucleic Acid Ther.
24: 313–325.
- 7. Berman, C.
L., Barros,
S. A.,
Galloway, S.
M., Kasper,
P., Oleson,
F. B.,
Priestley, C.
C., Sweder,
K. S.,
Schlosser, M.
J. and Sobol,
Z.
2016. OSWG recommendations for genotoxicity testing of novel
oligonucleotide-based therapeutics. Nucleic Acid
Ther.
26: 73–85.
- 8. Marlowe, J.
L., Akopian,
V.,
Karmali, P.,
Kornbrust,
D.,
Lockridge, J.
and Semple,
S.
2017. Recommendations of the oligonucleotide safety working
group’s formulated oligonucleotide subcommittee for the safety assessment of formulated
oligonucleotide-based therapeutics. Nucleic Acid
Ther.
27: 183–196.
- 9. MHLW Japan.
2020. Guidelines for Nonclinical Safety Assessment of Nucleic Acid
Pharmaceuticals. https://www.mhlw.go.jp/hourei/doc/tsuchi/T200331I0040.pdf [accessed April
30, 2020].
- 10. MHLW/PMDA Japan.
2012. Preclinical Safety Evaluation of Biotechnology-Derived
Pharmaceuticals S6(R1), https://www.pmda.go.jp/files/000156596.pdf [accessed April 30,
2020].
- 11. Geary, R.
S., Baker,
B. F. and
Crooke, S.
T.
2015. Clinical and preclinical pharmacokinetics and
pharmacodynamics of mipomersen (kynamro®): a second-generation antisense
oligonucleotide inhibitor of apolipoprotein B. Clin.
Pharmacokinet.
54: 133–146.
- 12. Geary, R.
S., Yu, R.
Z., Watanabe,
T., Henry,
S. P.,
Hardee, G.
E., Chappell,
A., Matson,
J., Sasmor,
H.,
Cummins, L.
and Levin, A.
A.
2003. Pharmacokinetics of a tumor necrosis factor-alpha
phosphorothioate 2′-O-(2-methoxyethyl) modified antisense oligonucleotide: comparison
across species. Drug Metab. Dispos.
31: 1419–1428.
- 13. Yu,
R. Z.,
Lemonidis, K.
M., Graham,
M. J.,
Matson, J.
E., Crooke,
R. M.,
Tribble, D.
L., Wedel, M.
K., Levin,
A. A. and
Geary, R.
S.
2009. Cross-species comparison of in vivo
PK/PD relationships for second-generation antisense oligonucleotides targeting
apolipoprotein B-100. Biochem.
Pharmacol.
77: 910–919.
- 14. MHLW/PMDA Japan.
2010. Non-Clinical Safety Studies for the Conduct of Human Clinical Trials
and Marketing Authorization for Pharmaceuticals M3(R2) https://www.pmda.go.jp/files/000156128.pdf [accessed April 30,
2020].
- 15. MHLW/PMDA Japan.
2006. Impurities in New Drug Substances Q3A(R2) https://www.pmda.go.jp/files/000156346.pdf [acce ssed April 30,
2020].
- 16. MHLW/PMDA Japan.
2006. Impurities in New Drug Substances Q3B(R2) https://www.pmda.go.jp/files/000156811. pdf [accessed April 30,
2020].
- 17. MHLW/PMDA Japan.
2015. Assesment and Control of DNA Reactive (Mutagenic) Impurities in
Pharmaceuticals to Limit Potential Carcinogenic Risk M7, https://www.pmda.go.jp/files/000208234.pdf [accessed April 30,
2020].
- 18. MHLW/PMDA Japan.
2006. Impurities: Guideline for Residual Solvents Q3C, https://www.pmda.go.jp/files/
000215510.pdf [accessed April 30, 2020].
- 19. MHLW/PMDA Japan.
2015. Guideline for Elemental Impurities Q3D, https://www.pmda.go.jp/files/000197758. pdf [accessed April 30,
2020].
