REVIEW
Histochemistry, Cytochemistry and Epigenetics
2022 Volume 55 Issue 1 Pages 1-7

Details
2022 Volume 55 Issue 1 Pages 1-7
Over the past few decades, many researchers have individually identified tumor-related genes, and have accumulated information on their basic research in a database. With the development of technology that can comprehensively test the expression status within a short time, oncogene panel testing has become attainable. On the other hand, changes in gene expression that do not depend on changes in base sequences, that is, epigenetics, or more comprehensively, epigenomes, are also highly involved in the development and progression of disease. Oncogene panel tests tend to focus on DNA base mutations such as point mutations, deletions, duplications, and chimera formation. Elucidation leads to correct interpretation of diseases and treatment choices, and we are in an era where integrated understanding of the genome and epigenome is indispensable. In this review, we make every effort to cover a wide range of knowledge, including data on histone protein modification, non-coding (nc)RNA and DNA methylation, and recent application trials for demonstrating epigenetic alterations in histologic and cytologic specimens. We hope this review will help marshal the knowledge accumulated by researchers involved in genomic and epigenomic studies.
In 1953, when Watson and Crick demonstrated that DNA constituted a double helix comprising four complementary, fundamental bases, A-adenine, T-thymine, G-guanine, and C-cytosine, a new era of molecular biology began [45]. Sequences of these bases are accurately replicated during cell division and transmitted to daughter cells as genetic information. Despite various notions such as “challenging the mystery of DNA base sequences invades the realm of God”, or “the present earth was created by extraterrestrial organisms with advanced technology that gifted DNA nucleic acids with specific base sequences to the earth experimentally”, the human genome project was launched with the goal of 2003, 50 years after Watson and Crick’s report, as the year when reading the entire base sequences of one human being would be achieved, and analyzing the code made from those four bases would disclose the origin of human beings. With a budget of 3 billion dollars and within a period of 12 years, the decoding of the entire human genome of 3,200 Mb was completed [9]. As an offspring of this huge research budget, various technologies related to genome analysis progressed during this period, and now the complete, comprehensive human genome and RNA expression analysis is attainable in a day. With these technical advancements, cancer genomic medicine that comprehensively examines genomic abnormalities of human diseases, especially malignant tumors, makes possible the selection of molecular-targeted therapeutic agents corresponding to the genetic abnormalities of individual tumors. Also, treatment policies are being examined based on genomic information obtained from histopathological specimens and patient blood analysis [5].
The entire base sequences of many individual organisms other than of humans have also been compiled into a database, much of which is open to the public on the Internet. The base sequences of various organisms that have been analyzed so far are nematodes (total length 103 Mb, approximately 20,000 genes), similarly, yellow-spotted flies (180 Mb, 14,700 genes), Daphnia (200 Mb, 31,000 genes), corn (2,200 Mb, 45,000 or more genes), rice (430 Mb, 45,000 genes), mouse (2,600 Mb, 22,000 genes), and humans (total length 3,200 Mb, and about 23,000 genes). The total number of bases per cell tends to increase in higher animals. Of the genes that produce proteins, the number in humans is about the same as in nematodes, which is less than that in Daphnia. Consequently, the mechanism of how gene expression is temporally and spatially regulated is more important than the number of genes that determine the complexity (uniqueness) of the species. When compared to a keyboard instrument such as the piano, organ, or accordion, it is not the number of keys (the number of genes) but the musical notes that prescribe the composition, strength, and length that determines the characteristics (uniqueness) of music such as classic, jazz, children’s songs, and folk songs. The term epigenetics can be described as musical notes that determine how genes (keys) are expressed (pressed). The iPS cells [40, 41] created by Professor Yamanaka, who won the Nobel Prize in Physiology or Medicine, revealed that once differentiated, cells can be traced back to the developmental stage. This phenomenon of reprogramming is equivalent to listening to music from the beginning by intentionally returning to the beginning of the composition from the middle of the performance; thus, research on epigenetics is of particular importance in tandem with its potential expectations for regenerative medicine.
Epigenetics, a concept originally proposed by British embryologist Conrad Hal Waddington (1905–1975), is a term coined about “all the controlled processes that start with genetic material and shape the final organism” [44]. Except for immunoglobulin gene rearrangement and polymorphism of gene repeat sites (copy number variation), only one fertilized egg, while retaining the original DNA sequence (sharing the same genome), builds up the whole complex body comprising a variety of cells. Moreover, cells whose differentiation is determined in a certain direction (mostly cells at the tissue stem cell level) inherit cell-specific differentiation traits even after cell division. These mechanisms are broadly defined as epigenetics. Thus, the term “epigenetics” is now used to include many different biological phenomena, partly because the definition, per se, is markedly comprehensive when defined. Ironically, it is said that the concept of epigenetics has become much broader and more ambiguous in the centerpiece of research in the post-genome era, especially that a huge budget was allocated from NIH to research on epigenetics in 2008, and many researchers applied for grants under the assumption that their work was somehow related to epigenetics [22]. The definition of the term “epigenetics” is, therefore, very difficult to understand even for authors involved in research in areas related to epigenetics because the meaning can vary depending on the individual researcher. In this review, “epigenetic control mechanism” is defined as “the expression mechanism of genes controlled by changes in chromatin structure while keeping the base sequence of DNA unchanged” (Fig. 1).

Epigenetic regulation (relation between chromatin structural changes and gene expression). Gene expression in the dissociated state of chromatin (upper right), is activated and, conversely, the gene is inactivated in the aggregated state (lower left). By undergoing such regulation at each site on the chromosome, cells with the same DNA structure exert diversity in time and space.
Three typical epigenetic control mechanisms effect regulation of gene expression by chromatin structural changes: (1) histone protein modification [39], (2) non-coding RNA [46], and (3) DNA methylation [23, 28, 47] (Fig. 2). The interaction among these three mechanisms, as shown in Fig. 1, aggregates or relaxes the chromatin structure to reduce or stop gene expression, or to enhance or start gene expression [35]. Regarding the modification of histone protein in (1), gene expression is regulated by aggregating or relaxing the chromatin structure of the region containing the histone protein, according to the modification or mutation of the amino acid, at its specific site [31]. Among various histones and their variants, histone H3.3 is particularly important because it can replace the preexisting histone H3 and thereby alter the chromatin structure of quiescent cells [2, 6]. When a genetic abnormality occurs in the genome, especially in the histone protein itself, the histone protein dysfunctions [3, 4]. These diseases are caused by genomic abnormalities that lead to epigenetic abnormalities. The use of specific antibodies against structural alterations of histone is being advocated in histopathological diagnosis, such as in confirming the diagnosis of giant cell tumor of bone [1]. The non-coding RNA (ncRNA) in (2) is, on the other hand, complicated and diverse, and many of its functions, still unknown, are exactly the focus of the state-of-the-art research. ncRNA is a general term for all RNAs except messenger RNA (mRNA), which is translated into protein. The mRNA that produces proteins is transcribed from only 2% of the DNA of the entire genome, and most of the RNA transcribed from the entire genome is ncRNA. Of these, in addition to transfer RNA (tRNA), ribosomal RNA (rRNA), classic ncRNA groups such as small nuclear RNA (snRNA) and small nucleolar RNA (snoRNA) that have been newly discovered in the last 20 years [32]. Also known are small functional RNA groups such as miRNAs and siRNAs as short as 20 bases, piRNAs as short as 30 bases and long ncRNA (lncRNA) groups of about 200 or more bases [19]. Surprisingly, lncRNA exists in as many as or in more than 23,000 human genes and brings (raises?) a paradigm shift in the post genome era [19]. Some lncRNA is involved in epigenetic regulation by associating with chromatin modifiers, regulating gene positional relations in the nucleus, and in DNA methylation [30]. Moreover, lncRNA has a tissue-specific expression pattern that regulates various biological phenomena such as cell differentiation and ontogeny through mRNA stability, splicing, and protein translational regulation [30]. It is becoming clear that abnormalities in lncRNA are involved in the onset and aggravation of many diseases including malignancies [11]. While the number of studies on lncRNA has recently been on the increase, some recent inconsistent information is attributed to a number of researchers reporting diametrically contradictory data and gainsaying one another. To make matters worse, paper-fabricating factories, termed “paper mills” [24], that sell myriad, forged manuscripts based on paper templates and reusable data for monetary gain, cast an unfavorable light on research in the ncRNA field. China has a market of about 500 million yen for such fabrications, and it is said that thousands of papers targeting fields such as mRNA and lncRNA have already been fabricated and sold by those “paper mills” [24]. Misrepresenting natural science papers by manipulating Western blot bands, bar graphs, etc., reusing them and creating swarms of fake research papers, the “mills” sell them to researchers and, at almost the same time, submit them to different journals [24]. Therefore, elucidating the precise functions of ncRNA will require a great deal of effort in expunging the fabrications from authentic research papers; we must bide our time for the forged papers to be exposed, eliminated and the “mills” extirpated.

Three elements of epigenetics. DNA methylation, histone protein modification, and ncRNA are fundamental factors that change the structure of chromatin. They influence each other and implement complicated epigenetic control.
Regarding (3), DNA methylation is considered to play a central role in transcriptional regulation through epigenetics [23]; its modification affects the formation and modification of nucleosomes and chromatin, and plays the role of a “switch” that determines gene on/off. DNA methylation functions by a mechanism that maintains the pattern of methylation even after cell division, which is one of the characteristics of epigenetics—the differentiation trait is inherited even after subsequent cell division. As fundamental matter of DNA methylation, four bases A, G, T, and C combine to form high molecular weight DNA. When two bases 5'-CG-3' are lined up, the sequence is usually called CpG. Cytosine at this site is often methylated at the 5-position carbon atom of the pyrimidine ring. Once cytosine has been methylated, while newly synthesized cytosine is at first unmethylated, soon after cell division, it will eventually be methylated by the action of a methyl group transfer enzyme called DNMT1 by the maintenance methylation mechanism [13]. As a consequence, a methyl group was rapidly added to the Cytosine of 3'-GC-5' in the DNA, and 5'-CmG-3' and 3'-GCm-5' were maintained and promptly paired (Fig. 3). Thus, the distribution pattern of methylated cytosine is maintained even after cell division, and plays the role of transmitting genetic information as a fifth base, other than A, G, T, and C. Unlike genomic information comprising A, G, T, and C, which is strictly protected by multiple mechanisms such as DNA repair and elimination mechanism through apoptosis, de novo methylation or demethylation occurs physiologically. The dual nature of sustainability and diversity of methylated cytosine, therefore, represents the epigenetic mechanism itself, which is essential for stable ontogeny and maintenance of diverse tissue differentiation.

Methylation maintained during DNA replication and cell division is transmitted to post-dividing cells. Unmethylated cytosine is shown in blue and methylated cytosine is shown in red. Immediately after replication, cytosine is methylated, but the maintenance methylation mechanism by DNMT1 enzyme, which recognizes the hemimethylated state after replication, works and is rapidly methylated.
Methylated cytosine present at the following sites in the genome is considered to possess functional significance.
Regulation of gene expression by methylated cytosine accumulated in CpG-islandThe CpG site, which has a high density of CpG sequences in the gene expression regulatory region near the transcription start site and accumulates in the 0.5 kb to 2 kb region (called CpG-island), is a regulator of gene expression [14]. When the gene is actively expressed, it is maintained in a hypomethylated state, and when methylation accumulates in this CpG-island (CpG-island hypermethylation), it inactivates the gene [8]. The methylated cytosine accumulated in CpG-island recruits methylated DNA-binding proteins (MBD1-4, MeCP2, etc.), histone protein modifiers, and chromatin remodeling factors, all of which form a complex at the site of methylated cytosine [10]. As a result of these sequential events, the chromatin structure is aggregated, and genes are inactivated as shown in Fig. 1. BANP (BTG3-associated nuclear protein), one of the proteins with a BEN domain, is responsible for the regulation of gene expression in CpG-island [8]. It cannot bind to DNA when methylated, that is, BANP belongs to one of the next mentioned methylation-sensitive transcriptional regulator group and functions as the most important factor that determines the activation and inactivation of CpG-island (Fig. 4) [8]. Physiologically, such regulation of gene expression by CpG-island methylation modification is involved in heterochromatin formation, X-chromatin inactivation, the genomic imprinting phenomena, and selective gene expression at the developmental stage [34]. On the other hand, from a pathological point of view, this regulation is involved in genomic imprinting abnormalities including uniparental disomy, tumor development processes of various benign [29] and malignant [25] tumors, and inactivation and reactivation of various genes in neuropsychiatric disorders [42]. Besides conventional genetic pathways including point mutations, chromosomal translocations, and loss of heterozygosity (LOH), a carcinogenic pathway based on epigenetic alterations is called the epigenetic pathway [43], which is drawing attention as a factor related to the diversity of tumor progression from early stage of carcinogenesis, and as a therapeutic target.
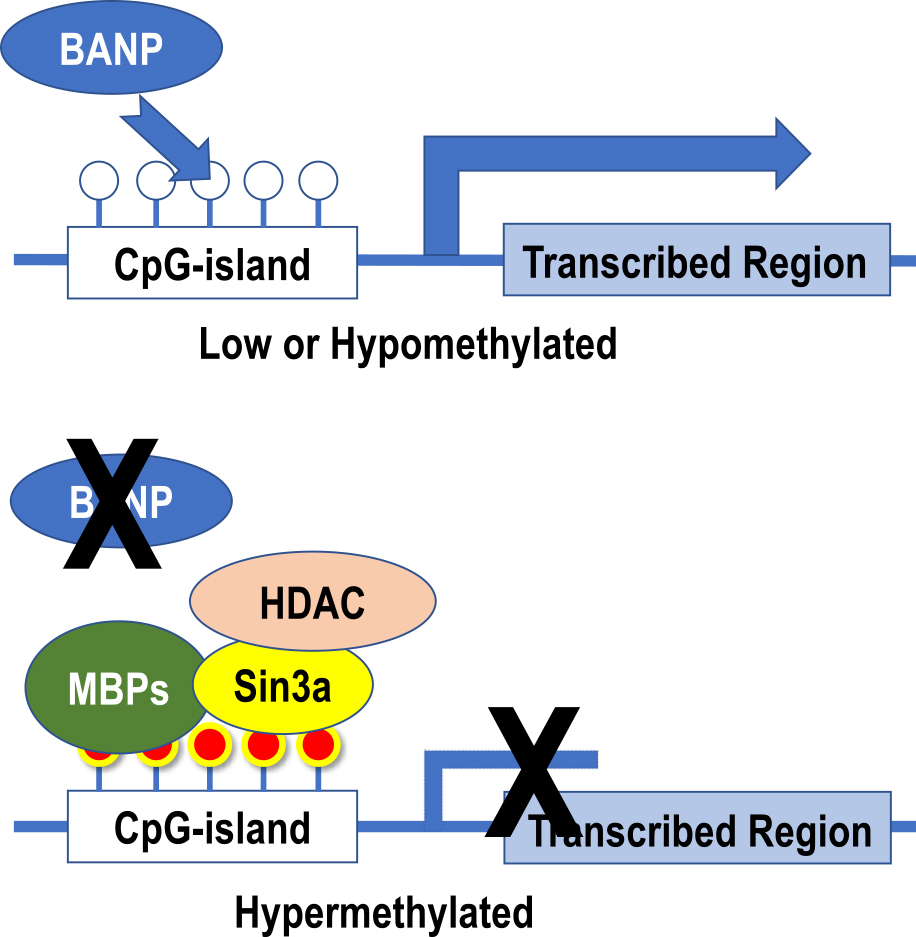
Changes in gene expression regulation by CpG-island methylation. When CpG-island is less methylated, BANP binds to DNA, allowing the chromatin structure to maintain its dissociated state and to effect transcription. On the other hand, when CpG-island becomes hypermethylated, BANP binding is inhibited. Instead, methylated cytosine-binding proteins (MBPs) recognize and bind to methylated cytosine, and Sin3a and histone deacetylase (HDAC) form complexes that alter the structure of surrounding histone proteins. Finally, the chromatin structure aggregates and gene expression ceases.
When methylation is present in CpG in the DNA binding sequence of a transcriptional regulator, the presence of a methyl group may prevent the transcriptional regulator from directly binding to DNA. In addition to the BANP [8] mentioned above, methylation-sensitive transcription factors such as Kaiso, AP-2, MYC, NF-κB, and ETS that are transcription factors related to systemic lupus erythematosus, fall under this category [37]. Sp1 and AP-1, usually considered to be methylation-insensitive transcriptional regulators, may also inhibit the binding of transcriptional regulators to DNA when methylation is present in CpG near the DNA-binding sequence of the transcriptional regulators [7, 17].
Methylated cytosine binding protein, MeCP2, mediatedTranscription may be suppressed by the methylation cytosine binding protein, MeCP2, occupying the binding site of the transcriptional regulator [27]. In particular, MeCP2 binds to a repeating sequence of 5 or more T/A and its adjacent methylated cytosine with high affinity [20]. Even a single incidence of methylation at the CpG site near TATA-box, which is one of the key points of gene expression regulation, may totally silence the expression without obvious hypermethylation of CpG-island [16, 26].
Of three major players in epigenetics, while immunohistochemistry uses specific antibodies for modified histone proteins, and conventional in situ hybridization is used for ncRNA, a technique for sequence-specific demonstration of methylated cytosine is still under development. Since tissue is composed of heterogeneous cell populations like tumor cells and surrounding stromal cells, we need to deal with highly selected cell populations for morphology-oriented epigenetic research, which is currently hardly cleared by microdissection. Recently, Koji et al. have introduced, for the first time, is an in situ technique termed HELMET: a reliable method for demonstrating sequence-specific DNA methylation at CCGG sites by comparing the digestion patterns of two isoschizomers, MspI and HpaII, of differing sensitivities to CpG methylation [21]. HELMET is, to date, the only reliable and effective method for demonstrating methylation at CCGG sites [23]. To establish a sequence-specific in situ demonstration of methylcytosine, we are currently developing a technique with the use of the padlock probe and rolling-circle amplification on tissue samples after bisulfite modification [18]. This amplification technique originally introduced by Nilsson et al. [33] is an alternative to conventional PCR. We also assayed an ICON-modified version of in situ demonstration of single methylated cytosine in a sequence-dependent manner to facilitated both specificity and sensitivity [18].
Epigenetics is a physiologically important mechanism that regulates cell development/differentiation and reprogramming of iPS cells, while its breakdown and temporal changes are involved in aging, known as chrono-epigenetics. It is becoming clear that elucidating and controlling the essence of epigenetics is a markedly important undertaking from the viewpoint of disease prevention and treatment, and of its use as a biomarker for epigenome diagnosis and treatment and, additionally in genomic medicine.
The leap in research on epigenetics over the last 12 years has been significant; in particular, knowledge has been accumulating on various modifications of histone proteins, their functional significance, and on diseases caused by abnormal histone structures. Moreover, epigenome analysis technology that comprehensively analyzes the methylation status of the entire genome has progressed to the extent that analyzing multiple cases of identical twins makes known which one develops a psychiatric disorder [15]. Epigenetic changes involved in the development of neuropsychiatric disorders are becoming clear [38]. Furthermore, epigenome analysis has brought about new developments in the effects of environmental factors on individuals and populations. Due to mental, physical, and sexual abuse suffered at the age of 3–5, DNA methylation occurs in a specific region, and the methylation pattern is preserved even after adulthood; also, environmental factors affect such individuals [15]. Environmental factors, recorded in the form of population DNA methylation patterns, are preserved across generations [12]. In that study, unknown genomic changes may have been involved in DNA methylation patterns at specific sites, and the simple concept of “epigenetics = inheritance of acquired traits” is a trait in genetics. This is a leap of logic and does not connote that Lysenko’s theory will be revived. Although recent data show the pattern memory of methylation brought about by environmental factors is inherited not only by the individual but also across generations, some of the acquired traits undergo molecular changes, to some extent, called epigenetics. It seems certain that the trait is transmitted through generations.
Finally, regarding methylated cytosines other than CpG-island (which actually make up most of the methylated cytosines present in cells) and, to a small extent, methylation modifications present in bases other than cytosine. Little is known about its function and significance [18, 36]. It is assumed that “atypical methylation” is involved in the regulation of lncRNA expression in some way, and its function is expected to be clarified as future research progresses.
Supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
The authors have no conflicts of interest.