Abstract
Neural stem/progenitor cells (NSPCs) in specific brain regions require precisely regulated metabolite production during critical development periods. Purines—vital components of DNA, RNA, and energy carriers like ATP and GTP—are crucial metabolites in brain development. Purine levels are tightly controlled through two pathways: de novo synthesis and salvage synthesis. Enzymes driving de novo pathway are assembled into a large multienzyme complex termed the “purinosome.” Here, we review purine metabolism and purinosomes as spatiotemporal regulators of neural development. Notably, around postnatal day 0 (P0) during mouse cortical development, purine synthesis transitions from the de novo pathway to the salvage pathway. Inhibiting the de novo pathway affects mTORC1 pathway and leads to specific forebrain malformations. In this review, we also explore the importance of protein-protein interactions of a newly identified NSPC protein—NACHT and WD repeat domain-containing 1 (Nwd1)—in purinosome formation. Reduced Nwd1 expression disrupts purinosome formation, impacting NSPC proliferation and neuronal migration, resulting in periventricular heterotopia. Nwd1 interacts directly with phosphoribosylaminoimidazole–succinocarboxamide synthetase (PAICS), an enzyme involved in de novo purine synthesis. We anticipate this review will be valuable for researchers investigating neural development, purine metabolism, and protein-protein interactions.
I. Purine Metabolism
Purines (compounds containing a pyrimidine ring fused with an imidazole ring) are found in all living organisms. They play essential roles as the building blocks of DNA and RNA, especially adenine and guanine, the energy molecules such as ATP and GTP, and the second messengers in both intracellular and extracellular signaling pathways such as cyclic AMP, cyclic GMP, adenosine and ATP [69]. Additionally, adenosine and ATP participate in extracellular signaling through purinergic receptors, which regulate various cell functions such as cell migration, apoptosis, proliferation, and differentiation [34, 43]. Figure 1 provides an overview of purine metabolism, illustrating the metabolites, enzymes, and chemical inhibitors. There are two pathways for purine synthesis in mammals: 1) the de novo purine synthesis and 2) the salvage purine synthesis (henceforth referred to as the de novo pathway and salvage pathway). In the salvage pathway, purines are synthesized by reusing degraded bases, including AMP, GMP, and IMP obtained from catabolism [36]. The enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT or HPRT) converts hypoxanthine and guanine to IMP and GMP, respectively, while adenine phosphoribosyltransferase (APRT) synthesizes AMP from adenine (Fig. 1) [56]. Under normal conditions, purines are predominantly supplied through the salvage pathway. However, the de novo pathway is activated when cellular demand for purines exceeds the capacity of salvage synthesis, particularly in situations that require higher levels of purines and their derivative nucleotides, such as during tumor growth and cell proliferation [55, 82].

The de novo pathway generates IMP from 5-phosphoribosyl-1-pyrophosphate (PRPP)—which is formed within the glycolysis-pentose phosphate pathway—through ten sequential reactions mediated by six enzymes [55, 56]. The six enzymes are: 1) phosphoribosyl pyrophosphate amidotransferase (PPAT); 2) glycinamide ribonucleotide transformylase (GART); 3) formylglycinamidine ribonucleotide synthase (FGAMS); 4) phosphoribosylaminoimidazole–succinocarboxamide synthetase (PAICS); 5) adenylosuccinate lyase (ADSL); and 6) 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase (ATIC) (Fig. 1). In addition to PRPP, aspartate from the tricarboxylic acid cycle, and glycine and formate from the folate biosynthesis pathway are required to drive the de novo pathway [55]. Formate—generated by the mitochondrial enzymes dihydrofolate reductase (DHFR), Serine hydroxymethyltransferase-2 (SHMT2), and methylenetetrahydrofolate dehydrogenase (MTHFD) as part of one-carbon metabolism—is converted to 10-Formyltetrahydrofolate (10-formyl-THF) and integrated into the de novo pathway through cytosolic enzymes GART and ATIC (Fig. 1) [19, 38, 55, 84].
II. Purinosome
In 2008, a significant advancement in purine metabolism was made with the discovery of the purinosome. The purinosome is a complex of six de novo pathway enzymes (PPAT, GART, FGAMS, PAICS, ADSL and ATIC). Assembled as a large multi-complex structure, it boosts the efficiency of de novo purine synthesis, particularly in cancer cells (Fig. 2A, B) [2, 56]. The purinosome is a metabolon, a complex structure comprising sequential metabolic enzymes [56]. Additionally, the purinosome contains several chaperones, including HSP70, HSP90, DnaJA1, and DnaJc7 [25, 57].

The molecular triggers for purinosome formation are not fully understood. The first three enzymes in the de novo pathway (tetrameric PPAT, monomeric FGAMS, and dimeric GART) form the purinosome core; the octameric PAICS, tetrameric ADSL, and dimeric ATIC, subsequently, interact with this core [29, 42, 56, 70]. By spatially organizing these enzymes, the purinosome minimizes the diffusion of intermediates and facilitates the rapid and coordinated synthesis of purine nucleotides. Pareek et al. demonstrated this through in situ three-dimensional sub-micrometer chemical imaging of single HeLa cells using gas cluster ion beam secondary ion mass spectrometry (GCIB-SIMS). They revealed that the purinosome is not merely a protein aggregate. It activates de novo purine synthesis, thus promoting the production of purine metabolites [55]. Furthermore, a recent study identified endogenous purinosome puncta using PAICS-GFP knock-in HeLa cells [12]. To ensure efficient purine metabolite production, purinosomes tend to form near the mitochondria, presumably to utilize the mitochondrial metabolites produced in the folate biosynthesis pathway [26]. In addition, timelapse imaging showed that purinosomes exhibit directed transport; they move along microtubules towards the mitochondria [3, 11].
Purinosome formation is regulated by various cellular signals and conditions, including substrate availability, energy status, and the cell cycle stage. Notably, purinosome formation is highest during the G1 phase of the cell cycle [10]. Cellular signaling pathways, such as those involving AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR), can influence purinosome assembly in response to changes in cellular energy levels [26, 45, 63]. Purinosomes can be induced to form in vitro by a) purine depletion using dialyzed fetal bovine serum (FBS) [2]; b) casein kinase II (CK2) inhibition using inhibitors such as DMAT and TBI [4]; c) stimulation of G protein-coupled receptors (GPCRs) with agonists like oxymetazoline [71]; d) disrupting salvage pathway (HGPRT knockdown) [27]; e) causing mitochondrial dysfunction using antimycin A and oligomycin [26]; f) inducing hypoxia [17]; and g) accelerating liquid-liquid phase separation (Fig. 2C) [12, 58].
III. Neural Development
The spatiotemporal balance of proliferation and differentiation of neural stem/progenitor cells (NSPCs) are crucial for the normal central nervous system (CNS) development. In the developing cerebral cortex of mammalian embryo, two distinct types of NSPCs are present: 1) the apical progenitors (APs) (also called apical radial glia) located in the apical-most part of the neocortex known as the ventricular zone, and 2) the basal progenitors (BPs) generated by APs, located in the subventricular zone. BPs consist of two major subtypes: basal intermediate progenitors (bIPs) and basal (outer) radial glia (bRG) [23, 31]. Primates, including humans, have abundant bRG, whereas rodents rarely have this subtype [31]. Newborn neurons generated from NSPCs in the ventricular and subventricular zones migrate radially toward the cortical plate and undergo sequential changes in cell shape. Defects in the proliferation of NPSCs and neuronal migration can lead to brain malformations such as microcephaly, lissencephaly, and periventricular heterotopia (PH), as well as contribute to various psychiatric disorders, including epilepsy and mental retardation [30, 61].
IV. Purine Metabolism in Neural Development
Unraveling purine metabolism is crucial for understanding brain development and its related abnormalities. In humans, several neurological pathologies are linked to purine metabolism. Inherited deficiencies in de novo pathway enzymes often lead to fetal lethality or neurological diseases [36]. For instance, a missense mutation in PAICS causes multiple physical malformations, including a small body, short neck, and craniofacial dysmorphism, and causes early neonatal death [59]. Moreover, deficiencies in ADSL or ATIC result in severe developmental brain defects such as mental retardation, autism, epilepsy, microcephaly, congenital blindness, speech impairment, and auto-aggressive behavior [20, 37, 49]. Similarly, prenatal exposure to methotrexate (MTX), which inhibits DHFR (a folate biosynthesis pathway enzyme) (Fig. 1), leads to fetal death. Moreover, children who survive this treatment, often exhibit cranial dysostosis, dysmorphic facial features, skeletal malformations, limb defects, growth retardation, and developmental delay; and they also exhibit severe CNS anomalies including semilobar holoprosencephaly (HPE), holoprosencephaly, and other brain malformations [13, 64]. Salvage pathway deficiencies, such as HGPRT deficiency, lead to Lesch–Nyhan syndrome, characterized by juvenile gout, dystonia, mental retardation, and compulsive self-injurious behavior [41, 68]. Previous studies of human pathologies strongly suggest the importance of a balance between de novo and salvage purine synthesis pathways for healthy brain development.
Our recent study delved into the expression profile and functional significance of purine synthesis enzymes in the developing mammalian brain. Each purine pathway exhibits distinct temporal and regional characteristics during brain development [50]. We revealed that purine synthesis transitions from the de novo to the salvage pathway in the neonatal period (Fig. 3A) [50]. FGMAS and PAICS, de novo pathway enzymes, are expressed abundantly in the early embryonic stages, while HGPRT expression is upregulated in the postnatal period (Fig. 3A). Hypoxanthine, a salvage pathway metabolite, influences morphologic changes in microglia [54]. Microglia likely represent the primary cell type utilizing the salvage pathway from late embryonic to postnatal stages. Immunohistochemistry—using anti-FGAMS, anti-PAICS, and anti-HGRPT antibodies in developing and adult mice brains—revealed that utilization of purine synthesis pathways during cerebral and cerebellar cortex development varies according to developmental stage and is region-specific [50]. Ultrastructural analysis using immunoelectron microscopy in primary cultured rat hippocampal neurons revealed that FGAMS, PAICS, and ATIC are localized in the mitochondria or near it [76]. In vitro study demonstrated that MMF (a de novo GMP synthesis inhibitor) but not forodesine (a salvage pathway inhibitor) significantly reduced NSPC proliferation and induced apoptosis (Fig. 3B) [50]. Similarly, multipotent NSPC proliferation relies solely on the active de novo pathway, whereas glial cell proliferation relies on either de novo or salvage pathways [50, 62]. Purinergic signaling is essential for NSPC maintenance and neuronal migration in the neocortical subventricular zone during brain development [43, 44]. In addition, purine metabolites, including ATP and GTP/GDP, are critical for polarity formation in postmitotic cortical neurons [60]. We found that PAICS knockdown (via in utero electroporation) reduces the NSPCs population and causes defects in neuronal migration during embryonic cortical development (Fig. 3C) [80]. Other studies using ADSL-knockdown chicken and zebrafish embryos revealed impaired neurogenesis and microcephaly accompanied by defective ciliogenesis [20]. Thus, both—reduced purine levels and impaired de novo purine synthesis—contribute to purine-related neurodevelopmental pathologies.

Through in vivo pharmacological studies using different inhibitors of purine synthesis, we confirmed that the spatiotemporal regulation of these two purine synthesis pathways is essential for normal mouse brain development. Experiments with mice treated with MMF, MTX, or forodesine showed that both pathways cooperatively regulate cerebellar development [41]. On the other hand, inhibitors targeting de novo pathway exclusively suppressed NSPC proliferation and delayed neuronal migration in the developing cerebral cortex [50]. Intriguingly, embryos continuously exposed to MMF exhibit severe forebrain cortical malformations, suggesting a gradient of purine demand along the anteroposterior axis of the embryonic brain, with higher requirements in the dorsal forebrain cortical areas than in the ventral or posterior areas (Fig. 3D, E) [50]. MMF-treated rostral neocortex expressed glutathione synthetase 2 (Gsh2–a marker of ganglionic eminence) but not paired box protein Pax6, suggesting that rostral neocortex is derived from the lateral ganglionic eminence (Fig. 3D, E) [50]. In addition to treatment with MMF, we showed that the treatments with MTX and LTX also induce severe brain malformations and neural tube defects [50, 73, 78].
De novo purine synthesis is also important for the proper functioning of the mature brain. FGAMS is weakly expressed in the adult brain [48, 50, 76]. Correspondingly, continuous MTX administration to mice (juvenile-age onwards) reduces adult neurogenesis in the hippocampus and causes deficits in spatial memory and visual recognition memory [74, 75, 83]. These findings highlight the continued requirement of de novo purine synthesis in specific brain regions and cell types even into adulthood. Thus, these findings contribute to our understanding of neurological disorders linked to de novo pathway defects.
In mammalian cultured cells, purine production is closely related to the mammalian/mechanistic target of the rapamycin complex 1 (mTORC1) signaling pathway and the expressions of eukaryotic initiation factor 4E (eIF4E)/binding protein 1 (4E-BP1) and ribosomal protein S6 kinase (S6K)/S6 proteins [8, 21, 33, 51]. Mutations in mTORC1 affect cerebrum development, suggesting its role in NSPCs development [5, 67]. We demonstrated that the administration of MHY1485 (an mTOR signal activator) [54] restores the MMF-induced cortical malformation [50]. Thus, the de novo pathway is strongly associated with mTORC1/S6K-S6 signaling cascade and may be related to mTORC1/4EBP1/eIF4E signaling cascade during cortical development [50].
Notably, the effects of deficiencies in the salvage and de novo pathways are different. HGPRT deficiency primarily leads to functional impairment of the dopamine system in the basal ganglia, while structural brain abnormalities are absent [32, 39, 72, 77]. Double knockout mice lacking both APRT and HGPRT (i.e., totally lacking salvage pathway) do not exhibit behavioral abnormalities related to Lesch–Nyhan syndrome [22]. Similarly, administration of forodesine to embryonic mice did not affect cortical development [50]. Thus, HGPRT deficiency mediates functional deficits in the dopamine system resulting from increased cell division and altered migration patterns of the midbrain dopamine neurons during early embryonic stages [77]. These in vivo findings might directly evidence the neurodevelopmental signature of Lesch–Nyhan syndrome. In vitro omics study using iPS lines from individuals with Lesch–Nyhan syndrome showed reductions in FAR2P1 mRNA, and NSEA4 and EID3 proteins, although the underlying reasons for these changes remain unclear [66]. Furthermore, cultured human NSPCs isolated from fetal brains of individuals with Lesch–Nyhan syndrome exhibited decreased FLT4 and β-tubulin III expression and impaired neuronal differentiation [15]. Further research is necessary to elucidate the molecular mechanisms of Lesch–Nyhan syndrome and to comprehensively interpret the in vitro and in vivo phenotypes associated with this syndrome.
V. Nwd1 in Brain Development
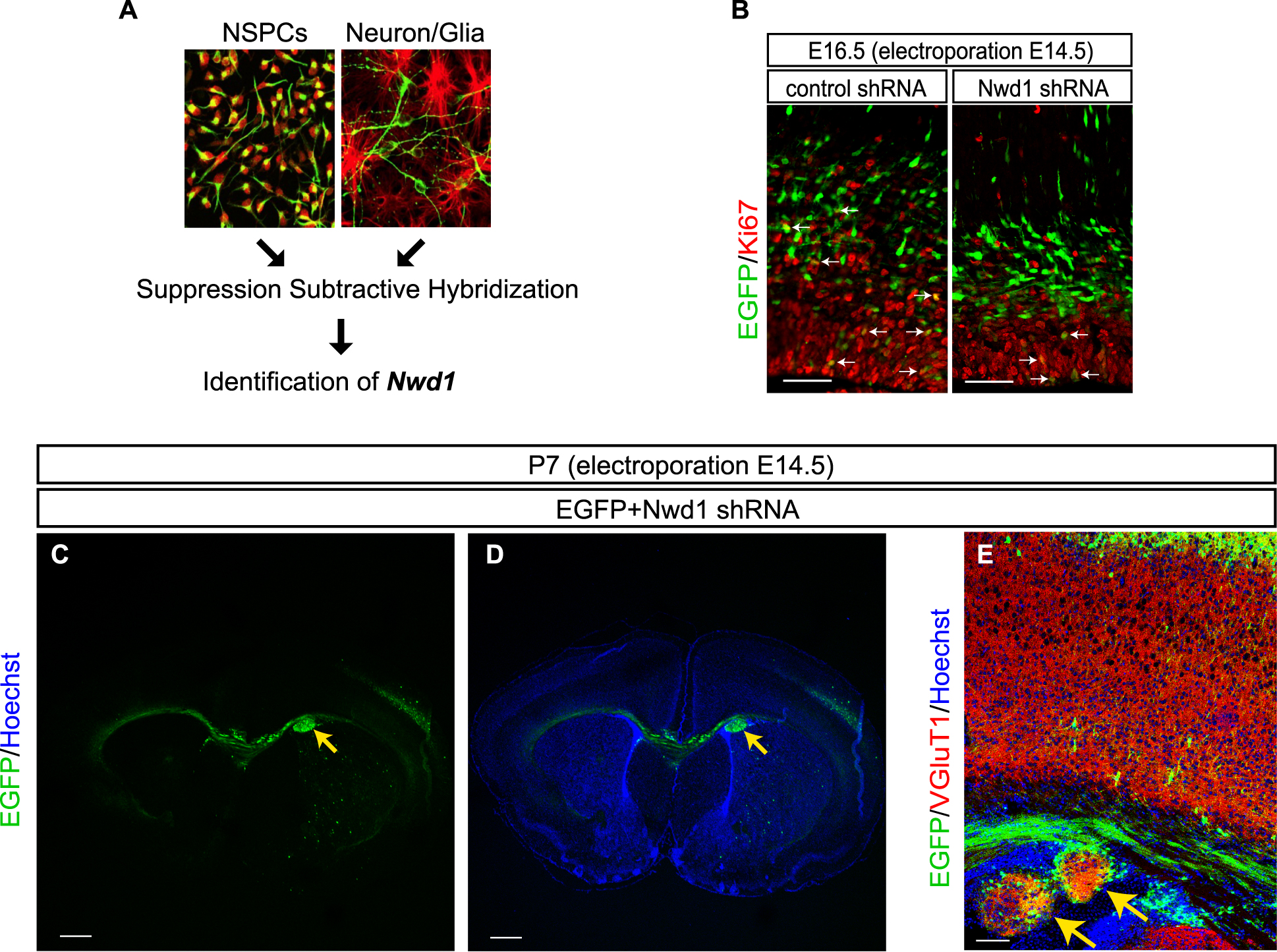
Previously, we employed suppression subtractive hybridization technique to identify novel genes highly expressed solely in NSPCs (Fig. 4A) [35, 79, 85]. This approach led us to discover the differentially expressed NACHT and WD repeat domain-containing protein 1 (Nwd1) gene; we confirmed robust expression of its transcript and protein in NSPCs during embryonic mouse brain development [79]. Nwd1 localizes in the mitochondria and endoplasmic reticulum [79, 81]. To elucidate the physiological roles of Nwd1, we suppressed Nwd1 expression (using in utero electroporation of Nwd1 shRNA) in the embryonic mouse cerebral cortex. Nwd1 knockdown resulted in defects in the Ki67+ proliferating NSPCs in the ventricular zone (Fig. 4B), premature differentiation into immature neurons, and delayed neuronal migration [80]. Notably, postnatal day 7 (P7) pups with suppressed Nwd1 expression at E14.5 exhibit PH, a developmental cortical dysgenesis frequently characterized by focal drug-resistant epilepsy in humans (Fig. 4C, D, Arrows) [7, 80]. PH is usually associated with an excessive formation of neural circuits by VgluT1+ excitatory glutamatergic neurons (Fig. 4E) [80].
VI. Nwd1 in Purinosome Assembly
The Nwd1 protein comprises a central NACHT domain (predicted to possess nucleoside-triphosphatase (NTPase) activity) and a cluster of WD40 repeats at the C-terminus [79]. Based on its molecular architecture, Nwd1 is classified as a member of the signal transduction ATPases with numerous domains (STAND) protein superfamily [79]. Other members of the STAND protein family often mediate ligand-induced self-oligomerization to form large multiprotein complexes such as the “apoptosome” and “inflammasome”; these complexes are induced by the apoptotic peptidase activating factor 1 (Apaf1) and nucleotide-binding oligomerization domain-like receptors (NLRs), respectively [9, 18, 40]. Notably, Nwd1 and Apaf1 have structurally analogous domains [18, 79]. While uncovering how Nwd1 regulates corticogenesis, we hypothesized that the N-terminal region of Nwd1 serves as an effector domain, enabling the protein to bind signaling molecules and trigger self-oligomerization mediated by its NACHT domain and WD40 repeats. Therefore, we sought to identify the Nwd1 binding partner, with a special focus on its N-terminal effector domain.
Various methodologies exist for studying protein-protein interaction (outlined in Table 1), each with its own strengths and limitations. The choice of method depends on specific research needs, including a) the type of interaction being studied, b) required sensitivity and specificity, c) whether the study is in vitro or in vivo, and 4) the available equipment and expertise. Among the available methods, we employed yeast-two-hybrid (Y2H) screening (TAKARA Bio) as a potent tool for identifying the unknown Nwd1 direct binding proteins (Fig. 5A) [80]. This approach led us to identify PAICS as the binding partner of the Nwd1-N terminal domain [80]. Further, we screened for proteins interacting with full-length Nwd1. Attempts to purify recombinant His- or GST-tagged Nwd1 protein using the bacterial expression system were unsuccessful due to insolubility issues (Fig. 5A). However, the Halo-tag-based binding assay using HEK293T cells and proteome analysis using LC-MS/MS proved effective in screening proteins interacting with full-length Nwd1 (Fig. 5A) [81]. A previous study expressing Flag-Nwd1 in LNCaP prostate cancer cell line identified multiple chaperones (such as HSP70 and HSP90) as full-length Nwd1 binding partners through co-immunoprecipitation and proteome analysis (Fig. 5A) [14].
Table 1.
Methods for detecting protein-protein interactions
|
Strengths |
Limitations |
|
・His-/GST-/MBP-/Halo-tag binding assay using E. coli and LC-MS/MS
|
・Low-cost
・High-purified-volume
・High specificity of purification
・Can be identified direct binding protein |
・Cannot be expressed over 100 kDa protein
・Easily insoluble
・May not reflect native interactions |
|
・Yeast-two-hybrid assay using Yeast
|
・Can be identified direct binding proteins
・Effective for screening |
・Cannot be expressed over 100 kDa protein
・Can be detected some false positives |
|
・Co-immunoprecipitation using mammalian cells and LC-MS/MS (antibody dependent)
|
・Any protein size
・Can be identified endogenously interacting protein |
・Can be detected indirectly interaction
・Dependent of antibody specification |
|
・Halo-tag binding assay using mammalian cells and LC-MS/MS (antibody independent)
|
・Any protein size
・Easily soluble
・High specificity of purification |
・Can be detected indirectly interaction
・low-purified-volume |
|
・FRET using mammalian cells and microscope
|
・Can be observed direct binding proteins
・High spatiotemporal resolution |
・Cannot be used for screening
・Need some specific equipment |
|
・Split fluorescent proteins using mammalian cells and microscope
|
・Can be observed direct protein binding
・High spatial resolution |
・Cannot be used for screening |
|
・Proximity ligation assay using mammalian cells and microscope
|
・Can be observed direct protein binding
・High spatial resolution |
・Cannot be used for screening |

Co-immunoprecipitation and immunostaining analysis also revealed Nwd1 as a novel member of purinosome alongside FGAMS and PAICS in HeLa cells (Fig. 5B) [80]. We showed that FGAMS and PAICS co-localize as granular structures in primary cultured NSPCs, indicating that purinosomes form within cellular processes and cell bodies even under normal physiological conditions (Fig. 5C) [80]. Our study was the first to evidence purinosome assembly under normal physiological conditions. Silencing the Nwd1 expression in primary cultured NSPCs reduces the formation of FGAMS+ and PAICS+ purinosomes, indicating that Nwd1 is involved in purinosome formation in NSPCs (Fig. 5D) [80]. In summary, our findings suggest that the dynamic assembly of purinosomes through the Nwd1-PAICS interaction is crucial for proper mammalian brain development (Fig. 5E).
Given that purinosomes are abundantly present in two cell types—tumor cells and NSPCs—with high purine demand, it is reasonable to assume that purinosomes also play a crucial role in stem cells of other tissues. However, direct evidence for in vivo localization of purinosome in various organs is currently lacking. Therefore, detecting purinosome formation and analyzing its functions in vivo will be crucial for future research. Moreover, the molecular factors responsible for the de novo to salvage pathway switch, and how de novo pathway inhibitors cause brain malformations, remain unclear. Future studies must focus on regional and lineage-specific variations in purine metabolism, especially in purine metabolites and related transcription factors in the brain. They will enhance our understanding of diseases resulting from abnormalities in purine metabolism, including defective purinosome formation. Such studies will also contribute to the development of anticancer medications which target purinosomes.
VII. Abbreviations
Metabolites: PRPP, 5-phosphoribosyl-1-pyrophosphate; 5-PRA, 5-phosphoribosylamine; GAR, glycinamide ribonucleotide; FGAR, formylglycinamide ribonucleotide; FGAM, formylglycinamidine ribonucleotide; AIR, aminoimidazole ribonucleotide; CAIR, 4-carboxy-5-aminoimidazole ribonucleotide; SAICAR, 4-(N-succinylcarboxamide) 5-aminoimidazole ribonucleotide; AICAR, aminoimidazole-4-carboxamide ribonucleotide; FAICAR, 5-formylaminoimidazole-4-carboxamide ribonucleotide; IMP, inosine monophosphate; XMP, xanthosine monophosphate; GMP, guanosine monophosphate; SAMP, adenylosuccinate; AMP, adenosine monophosphate; DHF, dihydrofolate; THF, tetrahydrofolic acid; ATP, adenosine triphosphate; GTP, guanosine triphosphate
Enzymes: PPAT, phosphoribosyl pyrophosphate amidotransferase; GART, phosphoribosylglycinamide formyltransferase; FGAMS, formylglycinamidine ribonucleotide synthase; PAICS, phosphoribosylaminoimidazole–succinocarboxamide synthetase; ADSL, adenylosuccinate lyase; ATIC, 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase; HGPRT, hypoxanthine-guanine phosphoribosyltransferase; APRT, adenine phosphoribosyltransferase; IMPDH, inosine monophosphate dehydrogenase; GMPS, GMP synthetase; 5'-NT, 5'-nucleotidase; PNP, purine nucleoside phosphorylase; ADSS, adenylosuccinate synthetase isozyme 2; ADA, adenosine deaminase; XO, xanthine oxidase; DHFR, dihydrofolate reductase; SHMT, Serine hydroxymethyltransferase, cytosolic; MTHFD1, methylenetetrahydrofolate dehydrogenase; MTR, 5-methyltetrahydrofolate-homocysteine methyltransferase
Chemical inhibitor: LTX, lometrexol; AG 2037, Pelitrexol; MRT, MRT00252040; MMF, mycophenolate mofetil; MZR, mizoribine; Forodesine, forodesine hydrochloride; DAP, 2,6-diaminopurine; MTX, methotrexate; SHIN1, SHMAT inhibitor 1; AZ, azathioprine; 6-MP, 6-mercaptopurine
VIII. Conflicts of Interest
The authors have no conflicts of interest to declare.
IX. Acknowledgments
This work was funded by the Japan Society for the Promotion of Science grants-in-aid (KAKENHI) grant numbers 21K20701 (to S. Y.) and 19K06931 (to S. S.). Gout and Uric Acid Research Foundation 2020 (to S. S), 2022 (to S. Y.), 2023 (to S. S.), and Waseda University Grants for Special Research Projects 2014K-6217 and 2015K-249 (to S. S.) 2021C-611 (to S. Y.). We also thank Enago (www.enago.jp) for English editing.
X. References
- 1 Allison, A. (2005) Mechanisms of action of mycophenolate mofetil. Lupus 14; 2–8.
- 2 An, S., Kumar, R., Sheets, E. D. and Benkovic, S. J. (2008) Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 320; 103–106.
- 3 An, S., Deng, Y., Tomsho, J. W., Kyoung, M. and Benkovic, S. J. (2010) Microtubule-assisted mechanism for functional metabolic macromolecular complex formation. Proc. Natl. Acad. Sci. U S A 107; 12872–12876.
- 4 An, S., Kyoung, M., Allen, J. J., Shokat, K. M. and Benkovic, S. J. (2010) Dynamic regulation of a metabolic multienzyme complex by protein kinase CK2. J. Biol. Chem. 285; 11093–11099.
- 5 Andrews, M. G., Subramanian, L. and Kriegstein, A. R. (2020) mTOR signaling regulates the morphology and migration of outer radial glia in developing human cortex. Elife 9; e58737.
- 6 Bailly, C. and Waring, M. J. (1998) The use of diaminopurine to investigate structural properties of nucleic acids and molecular recognition between ligands and DNA. Nucleic Acids Res. 26; 4309–4314.
- 7 Battaglia, G., Chiapparini, L., Franceschetti, S., Freri, E., Tassi, L., Bassanini, S., et al. (2006) Periventricular nodular heterotopia: classification, epileptic history, and genesis of epileptic discharges. Epilepsia 47; 86–97.
- 8 Ben-Sahra, I., Hoxhaj, G., Ricoult, S. J., Asara, J. M. and Manning, B. D. (2016) mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351; 728–733.
- 9 Cai, X., Xu, H. and Chen, Z. J. (2017) Prion-like polymerization in immunity and inflammation. Cold Spring Harb. Perspect. Biol. 9; a023580.
- 10 Chan, C. Y., Zhao, H., Pugh, R. J., Pedley, A. M., French, J., Jones, S. A., et al. (2015) Purinosome formation as a function of the cell cycle. Proc. Natl. Acad. Sci. U S A 112; 1368–1373.
- 11 Chan, C. Y., Pedley, A. M., Kim, D., Xia, C., Zhuang, X. and Benkovic, S. J. (2018) Microtubule-directed transport of purine metabolons drives their cytosolic transit to mitochondria. Proc. Natl. Acad. Sci. U S A 115; 13009–13014.
- 12 Chou, M. C., Wang, Y. H., Chen, F. Y., Kung, C. Y., Wu, K. P., Kuo, J. C., et al. (2023) PAICS ubiquitination recruits UBAP2 to trigger phase separation for purinosome assembly. Mol. Cell 83; 4123–4140. e4112.
- 13 Corona‐Rivera, J. R., Rea‐Rosas, A., Santana‐Ramírez, A., Acosta‐León, J., Hernández‐Rocha, J. and Miguel‐Jiménez, K. (2010) Holoprosencephaly and genitourinary anomalies in fetal methotrexate syndrome. Am. J. Med. Genet. A 152; 1741–1746.
- 14 Correa, R. G., Krajewska, M., Ware, C. F., Gerlic, M. and Reed, J. C. (2014) The NLR-related protein NWD1 is associated with prostate cancer and modulates androgen receptor signaling. Oncotarget 5; 1666–1682.
- 15 Cristini, S., Navone, S., Canzi, L., Acerbi, F., Ciusani, E., Hladnik, U., et al. (2010) Human neural stem cells: a model system for the study of Lesch–Nyhan disease neurological aspects. Hum. Mol. Genet. 19; 1939–1950.
- 16 Cronstein, B. N. (1997) The mechanism of action of methotrexate. Rheum. Dis. Clin. North Am. 23; 739–755.
- 17 Doigneaux, C., Pedley, A. M., Mistry, I. N., Papayova, M., Benkovic, S. J. and Tavassoli, A. (2020) Hypoxia drives the assembly of the multienzyme purinosome complex. J. Biol. Chem. 295; 9551–9566.
- 18 Dorstyn, L., Akey, C. W. and Kumar, S. (2018) New insights into apoptosome structure and function. Cell Death Differ. 25; 1194–1208.
- 19 Ducker, G. S. and Rabinowitz, J. D. (2017) One-carbon metabolism in health and disease. Cell Metab. 25; 27–42.
- 20 Dutto, I., Gerhards, J., Herrera, A., Souckova, O., Škopová, V., Smak, J. A., et al. (2022) Pathway-specific effects of ADSL deficiency on neurodevelopment. Elife 11; e70518.
- 21 Emmanuel, N., Ragunathan, S., Shan, Q., Wang, F., Giannakou, A., Huser, N., et al. (2017) Purine nucleotide availability regulates mTORC1 activity through the Rheb GTPase. Cell Rep. 19; 2665–2680.
- 22 Engle, S. J., Womer, D. E., Davies, P. M., Boivin, G., Sahota, A., Simmonds, H. A., et al. (1996) HPRT-APRT-deficient mice are not a model for Lesch-Nyhan syndrome. Hum. Mol. Genet. 5; 1607–1610.
- 23 Englund, C., Fink, A., Lau, C., Pham, D., Daza, R. A., Bulfone, A., et al. (2005) Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J. Neurosci. 25; 247–251.
- 24 Evans, G. B., Tyler, P. C. and Schramm, V. L. (2018) Immucillins in infectious diseases. ACS Infect. Dis. 4; 107–117.
- 25 French, J. B., Zhao, H., An, S., Niessen, S., Deng, Y., Cravatt, B. F., et al. (2013) Hsp70/Hsp90 chaperone machinery is involved in the assembly of the purinosome. Proc. Natl. Acad. Sci. U S A 110; 2528–2533.
- 26 French, J. B., Jones, S. A., Deng, H., Pedley, A. M., Kim, D., Chan, C. Y., et al. (2016) Spatial colocalization and functional link of purinosomes with mitochondria. Science 351; 733–737.
- 27 Fu, R., Sutcliffe, D., Zhao, H., Huang, X., Schretlen, D. J., Benkovic, S., et al. (2015) Clinical severity in Lesch–Nyhan disease: The role of residual enzyme and compensatory pathways. Mol. Genet. Metab. 114; 55–61.
- 28 Gearry, R. B. and Barclay, M. L. (2005) Azathioprine and 6‐mercaptopurine pharmacogenetics and metabolite monitoring in inflammatory bowel disease. J. Gastroenterol. Hepatol. 20; 1149–1157.
- 29 Greasley, S. E., Horton, P., Ramcharan, J., Beardsley, G. P., Benkovic, S. J. and Wilson, I. A. (2001) Crystal structure of a bifunctional transformylase and cyclohydrolase enzyme in purine biosynthesis. Nat. Struct. Biol. 8; 402–406.
- 30 Hansen, A. H., Duellberg, C., Mieck, C., Loose, M. and Hippenmeyer, S. (2017) Cell polarity in cerebral cortex development—cellular architecture shaped by biochemical networks. Front. Cell. Neurosci. 11; 176.
- 31 Hansen, D. V., Lui, J. H., Parker, P. R. and Kriegstein, A. R. (2010) Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464; 554–561.
- 32 Hooper, M., Hardy, K., Handyside, A., Hunter, S. and Monk, M. (1987) HPRT-deficient (Lesch–Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature 326; 292–295.
- 33 Hoxhaj, G., Hughes-Hallett, J., Timson, R. C., Ilagan, E., Yuan, M., Asara, J. M., et al. (2017) The mTORC1 signaling network senses changes in cellular purine nucleotide levels. Cell Rep. 21; 1331–1346.
- 34 Huang, Z., Xie, N., Illes, P., Di Virgilio, F., Ulrich, H., Semyanov, A., et al. (2021) From purines to purinergic signalling: molecular functions and human diseases. Signal Transduct. Target. Ther. 6; 162.
- 35 Iwasaki, Y., Yumoto, T. and Sakakibara, S. (2015) Expression profiles of inka2 in the murine nervous system. Gene Expr. Patterns 19; 83–97.
- 36 Jurecka, A. (2009) Inborn errors of purine and pyrimidine metabolism. J. Inherit. Metab. Dis. 32; 247–263.
- 37 Jurecka, A., Zikanova, M., Kmoch, S. and Tylki-Szymańska, A. (2015) Adenylosuccinate lyase deficiency. J. Inherit. Metab. Dis. 38; 231–242.
- 38 Kory, N., Wyant, G. A., Prakash, G., Uit de Bos, J., Bottanelli, F., Pacold, M. E., et al. (2018) SFXN1 is a mitochondrial serine transporter required for one-carbon metabolism. Science 362; eaat9528.
- 39 Kuehn, M. R., Bradley, A., Robertson, E. J. and Evans, M. J. (1987) A potential animal model for Lesch–Nyhan syndrome through introduction of HPRT mutations into mice. Nature 326; 295–298.
- 40 Leipe, D. D., Koonin, E. V. and Aravind, L. (2004) STAND, a class of P-loop NTPases including animal and plant regulators of programmed cell death: multiple, complex domain architectures, unusual phyletic patterns, and evolution by horizontal gene transfer. J. Mol. Biol. 343; 1–28.
- 41 Lesch, M. and Nyhan, W. L. (1964) A familial disorder of uric acid metabolism and central nervous system function. Am. J. Med. 36; 561–570.
- 42 Li, S. X., Tong, Y. P., Xie, X. C., Wang, Q. H., Zhou, H. N., Han, Y., et al. (2007) Octameric structure of the human bifunctional enzyme PAICS in purine biosynthesis. J. Mol. Biol. 366; 1603–1614.
- 43 Lin, J. H. C., Takano, T., Arcuino, G., Wang, X., Hu, F., Darzynkiewicz, Z., et al. (2007) Purinergic signaling regulates neural progenitor cell expansion and neurogenesis. Developmental Biology. 302; 356–366.
- 44 Liu, X., Hashimoto-Torii, K., Torii, M., Haydar, T. F. and Rakic, P. (2008) The role of ATP signaling in the migration of intermediate neuronal progenitors to the neocortical subventricular zone. Proc. Natl. Acad. Sci. U S A 105; 11802–11807.
- 45 Ma, E. H. and Jones, R. G. (2016) (TORC) ing up purine biosynthesis. Science 351; 670–671.
- 46 Makino, Y., Oe, C., Iwama, K., Suzuki, S., Nishiyama, A., Hasegawa, K., et al. (2022) Serine hydroxymethyltransferase as a potential target of antibacterial agents acting synergistically with one-carbon metabolism-related inhibitors. Commun. Biol. 5; 619.
- 47 Maltzman, J. S. and Koretzky, G. A. (2003) Azathioprine: old drug, new actions. J. Clin. Invest. 111; 1122–1124.
- 48 Mangold, C. A., Yao, P. J., Du, M., Freeman, W. M., Benkovic, S. J. and Szpara, M. L. (2018) Expression of the purine biosynthetic enzyme phosphoribosyl formylglycinamidine synthase in neurons. J. Neurochem. 144; 723–735.
- 49 Marie, S., Heron, B., Bitoun, P., Timmerman, T., Van den Berghe, G. and Vincent, M. F. (2004) AICA-ribosiduria: a novel, neurologically devastating inborn error of purine biosynthesis caused by mutation of ATIC. Am. J. Hum. Genet. 74; 1276–1281.
- 50 Mizukoshi, T., Yamada, S. and Sakakibara, S. (2023) Spatiotemporal Regulation of De Novo and Salvage Purine Synthesis during Brain Development. eNeuro 10; ENEURO.0159–23.2023.
- 51 Morita, M., Gravel, S. P., Chenard, V., Sikström, K., Zheng, L., Alain, T., et al. (2013) mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 18; 698–711.
- 52 Nakajo, A., Khoshnoodi, J., Takenaka, H., Hagiwara, E., Watanabe, T., Kawakami, H., et al. (2007) Mizoribine corrects defective nephrin biogenesis by restoring intracellular energy balance. J. Am. Soc. Nephrol. 18; 2554–2564.
- 53 Nomura, J., Busso, N., Ives, A., Tsujimoto, S., Tamura, M., So, A., et al. (2013) Febuxostat, an inhibitor of xanthine oxidase, suppresses lipopolysaccharide-induced MCP-1 production via MAPK phosphatase-1-mediated inactivation of JNK. PLoS One 8; e75527.
- 54 Okajima, T., Gu, Y., Teruya, R., Yano, S., Taketomi, T., Sato, B., et al. (2020) Atypical cadherin FAT3 is a novel mediator for morphological changes of microglia. eNeuro 7; ENEURO.0056–20.2020.
- 55 Pareek, V., Tian, H., Winograd, N. and Benkovic, S. J. (2020) Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science 368; 283–290.
- 56 Pedley, A. M. and Benkovic, S. J. (2017) A new view into the regulation of purine metabolism: the purinosome. Trends Biochem. Sci. 42; 141–154.
- 57 Pedley, A. M., Karras, G. I., Zhang, X., Lindquist, S. and Benkovic, S. J. (2018) Role of HSP90 in the regulation of de novo purine biosynthesis. Biochemistry 57; 3217–3221.
- 58 Pedley, A. M., Boylan, J. P., Chan, C. Y., Kennedy, E. L., Kyoung, M. and Benkovic, S. J. (2022) Purine biosynthetic enzymes assemble into liquid-like condensates dependent on the activity of chaperone protein HSP90. J. Biol. Chem. 298; 101845.
- 59 Pelet, A., Skopova, V., Steuerwald, U., Baresova, V., Zarhrate, M., Plaza, J. M., et al. (2019) PAICS deficiency, a new defect of de novo purine synthesis resulting in multiple congenital anomalies and fatal outcome. Hum. Mol. Genet. 28; 3805–3814.
- 60 Raman, R., Pinto, C. S. and Sonawane, M. (2018) Polarized organization of the cytoskeleton: regulation by cell polarity proteins. J. Mol. Biol. 430; 3565–3584.
- 61 Represa, A. (2019) Why malformations of cortical development cause epilepsy. Front. Neurosci. 13; 250.
- 62 Sato, K., Kanno, J., Tominaga, T., Matsubara, Y. and Kure, S. (2006) De novo and salvage pathways of DNA synthesis in primary cultured neurall stem cells. Brain Res. 1071; 24–33.
- 63 Schmitt, D. L., Cheng, Y. J., Park, J. and An, S. (2016) Sequestration-mediated downregulation of de novo purine biosynthesis by AMPK. ACS Chem. Biol. 11; 1917–1924.
- 64 Seidahmed, M. Z., Shaheed, M. M., Abdulbasit, O. B., Al Dohami, H., Babiker, M., Abdullah, M. A., et al. (2006) A case of methotrexate embryopathy with holoprosencephaly, expanding the phenotype. Birth Defects Res. A Clin. Mol. Teratol. 76; 138–142.
- 65 Sekine, M., Okamoto, K., Pai, E. F., Nagata, K., Ichida, K., Hille, R., et al. (2023) Allopurinol and oxypurinol differ in their strength and mechanisms of inhibition of xanthine oxidoreductase. J. Biol. Chem. 299; 105189.
- 66 Sutcliffe, D. J., Dinasarapu, A. R., Visser, J. E., Hoed, J. D., Seifar, F., Joshi, P., et al. (2021) Induced pluripotent stem cells from subjects with Lesch-Nyhan disease. Sci. Rep. 11; 8523.
- 67 Tarkowski, B., Kuchcinska, K., Blazejczyk, M. and Jaworski, J. (2019) Pathological mTOR mutations impact cortical development. Hum. Mol. Genet. 28; 2107–2119.
- 68 Torres, R. J. and Puig, J. G. (2007) Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome. Orphanet J. Rare Dis. 2; 1–10.
- 69 Traut, T. W. (1994) Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140; 1–22.
- 70 Vergis, J. M., Bulock, K. G., Fleming, K. G. and Beardsley, G. P. (2001) Human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase/Inosine 5'-monophosphate cyclohydrolase: a bifunctional protein requiring dimerization for transformylase activity but not for cyclohydrolase activity. J. Biol. Chem. 276; 7727–7733.
- 71 Verrier, F., An, S., Ferrie, A. M., Sun, H., Kyoung, M., Deng, H., et al. (2011) GPCRs regulate the assembly of a multienzyme complex for purine biosynthesis. Nat. Chem. Biol. 7; 909–915.
- 72 Visser, J., Bär, P. and Jinnah, H. (2000) Lesch–Nyhan disease and the basal ganglia. Brain Res. Brain Res. Rev. 32; 449–475.
- 73 Wang, J., Wang, X., Guan, T., Xiang, Q., Wang, M., Zhang, Z., et al. (2014) Analyses of copy number variation reveal putative susceptibility loci in MTX‐induced mouse neural tube defects. Dev. Neurobiol. 74; 877–893.
- 74 Welbat, J. U., Naewla, S., Pannangrong, W., Sirichoat, A., Aranarochana, A. and Wigmore, P. (2020) Neuroprotective effects of hesperidin against methotrexate-induced changes in neurogenesis and oxidative stress in the adult rat. Biochem. Pharmacol. 178; 114083.
- 75 Wen, J., Maxwell, R. R., Wolf, A. J., Spira, M., Gulinello, M. E. and Cole, P. D. (2018) Methotrexate causes persistent deficits in memory and executive function in a juvenile animal model. Neuropharmacology 139; 76–84.
- 76 Williamson, J., Petralia, R. S., Wang, Y. X., Mattson, M. P. and Yao, P. J. (2017) Purine biosynthesis enzymes in hippocampal neurons. Neuromolecular Med. 19; 518–524.
- 77 Witteveen, J., Loopstok, S., Ballesteros, L. L., Boonstra, A., van Bakel, N., van Boekel, W., et al. (2022) HGprt deficiency disrupts dopaminergic circuit development in a genetic mouse model of Lesch–Nyhan disease. Cell. Mol. Life Sci. 79; 341.
- 78 Xu, L., Wang, L., Wang, J., Zhu, Z., Chang, G., Guo, Y., et al. (2016) The effect of inhibiting glycinamide ribonucleotide formyl transferase on the development of neural tube in mice. Nutr. Metab. (Lond) 13; 1–10.
- 79 Yamada, S. and Sakakibara, S. (2018) Expression profile of the STAND protein Nwd1 in the developing and mature mouse central nervous system. J. Comp. Neurol. 526; 2099–2114.
- 80 Yamada, S., Sato, A. and Sakakibara, S. (2020) Nwd1 regulates neuronal differentiation and migration through purinosome formation in the developing cerebral cortex. iScience 23; 101058.
- 81 Yamada, S., Nakadate, K., Mizukoshi, T., Kawakami, K., Kobayashi, R., Horii, T., et al. (2024) Induction of NASH in the Nwd1−/− mouse liver via SERCA2-dependent endoplasmic reticulum stress. bioRxiv. 2024.2001.2026.577307.
- 82 Yamaoka, T., Kondo, M., Honda, S., Iwahana, H., Moritani, M., Ii, S., et al. (1997) Amidophosphoribosyltransferase limits the rate of cell growth-linked de novo purine biosynthesis in the presence of constant capacity of salvage purine biosynthesis. J. Biol. Chem. 272; 17719–17725.
- 83 Yang, M., Kim, J. S., Kim, J., Kim, S. H., Kim, J. C., Kim, J., et al. (2011) Neurotoxicity of methotrexate to hippocampal cells in vivo and in vitro. Biochem. Pharmacol. 82; 72–80.
- 84 Yang, M. and Vousden, K. H. (2016) Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 16; 650–662.
- 85 Yumoto, T., Nakadate, K., Nakamura, Y., Sugitani, Y., Sugitani-Yoshida, R., Ueda, S., et al. (2013) Radmis, a novel mitotic spindle protein that functions in cell division of neural progenitors. PloS One 8; e79895.
