Abstract
Protein functions can be predicted based on their three-dimensional structures. However, many multidomain proteins have unstable structures, making it difficult to determine the whole structure in biological experiments. Additionally, multidomain proteins are often decomposed and identified based on their domains, with the structure of each domain often found in public databases. Recent studies have advanced structure prediction methods of multidomain proteins through computational analysis. In existing methods, proteins that serve as templates are used for three-dimensional structure prediction. However, when no protein template is available, the accuracy of the prediction is decreased. This study was conducted to predict the structures of multidomain proteins without the need for whole structure templates.
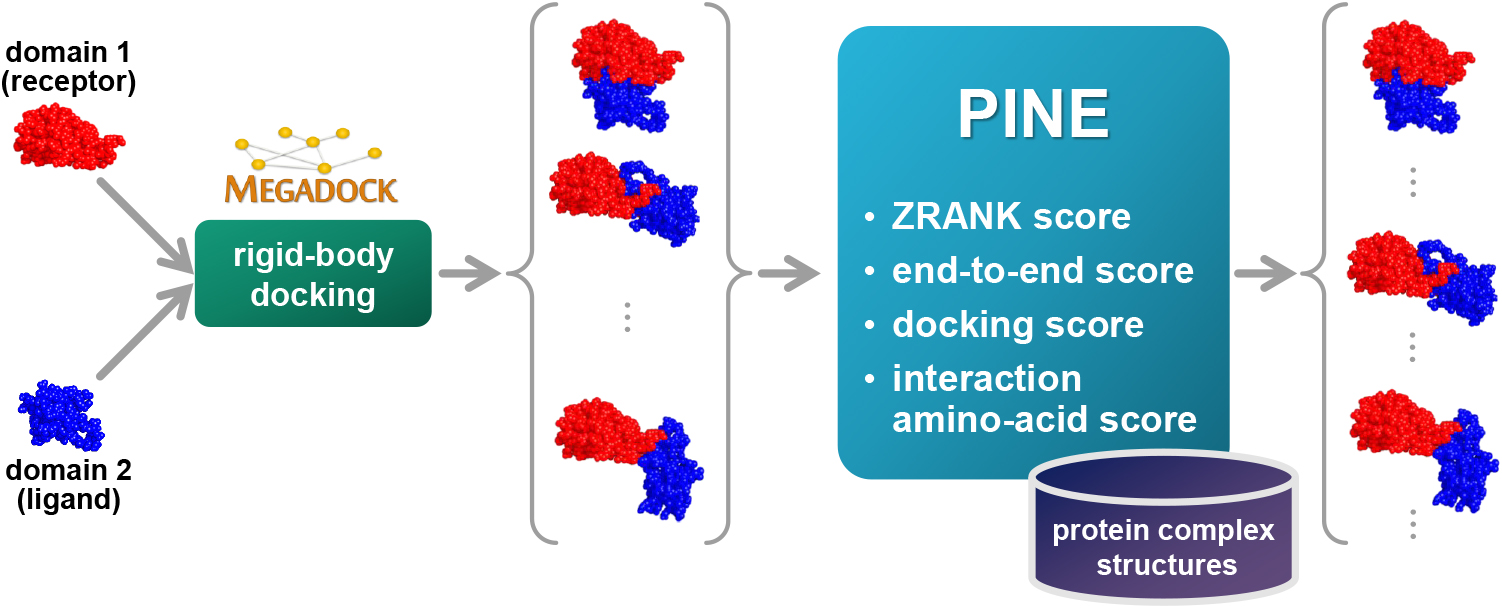
We improved structure prediction methods by performing rigid-body docking from the structure of each domain and reranking a structure closer to the correct structure to have a higher value. In the proposed method, the score for the domain-domain interaction obtained without a structural template of the multidomain protein and score for the three-dimensional structure obtained during docking calculation were newly incorporated into the score function. We successfully predicted the structures of 50 of 55 multidomain proteins examined in the test dataset.
Interaction residue pair information of the protein-protein complex interface contributes to domain reorganizations even when a structural template for a multidomain protein cannot be obtained. This approach may be useful for predicting the structures of multidomain proteins with important biochemical functions.