Review Article
Effect of nuclear import receptors on liquid–liquid phase separation
2020 Volume 17 Pages 25-29

Details
2020 Volume 17 Pages 25-29
Low-complexity (LC) sequences, regions that are predominantly made up of limited amino acids, are often observed in eukaryotic nuclear proteins. The role of these LC sequences has remained unclear for decades. Recent studies have shown that LC sequences are important in the formation of membrane-less organelles via liquid–liquid phase separation (LLPS). The RNA binding protein, fused in sarcoma (FUS), is the most widely studied of the proteins that undergo LLPS. It forms droplets, fibers, or hydrogels using its LC sequences. The N-terminal LC sequence of FUS is made up of Ser, Tyr, Gly, and Gln, which form a labile cross-β polymer core while the C-terminal Arg-Gly-Gly repeats accelerate LLPS. Normally, FUS localizes to the nucleus via the nuclear import receptor karyopherin β2 (Kapβ2) with the help of its C-terminal proline-tyrosine nuclear localization signal (PY-NLS). Recent findings revealed that Kapβ2 blocks FUS mediated LLPS, suggesting that Kapβ2 is not only a transport protein but also a chaperone which regulates LLPS during the formation of membrane-less organelles. In this review, we discuss the effects of the nuclear import receptors on LLPS.
Liquid–liquid phase separation (LLPS) is believed to be a fundamental mechanism during the assembly of membrane-less organelles including RNA granules. Low-complexity (LC) sequences of component proteins have been shown to make a significant contribution to LLPS. The advantage of membrane-less organelles formed by LLPS is flexibility that allows quick assembling and disassembling responding to diverse cellular contexts. Nuclear import receptors may play an important regulatory role in LLPS inhibition. Chaperone function of nuclear import receptors is thought to depend on the tight specific binding of these proteins to the nuclear localization signal and weak transient binding of the LC sequences.
The exchange of biological macromolecules between the cytoplasm and the nucleus occurs via the nuclear pores and is essential for their precise localization. Proteins and nucleic acids that are larger than 40 kDa cannot pass through the nuclear pores without the assistance of nuclear import receptors. These receptors, also known as Karyopherin β family proteins (Kapβs), mediate the active transport of these large macromolecules [1–3] to and from the nucleus. Much of the cargo imported by Kapβs are rich in intrinsically disordered low complexity (LC) sequences. For a long time, the role of these LC sequences was largely unknown. In the last ten years, liquid–liquid phase separation (LLPS) has been widely analyzed and several reviews on LLPS have been published [4–9]. Many groups have demonstrated that LC sequences/domains are important for LLPS [10–14]. Study of the LLPS phenomena has shed new light on the probable function of LC sequences in intrinsically disordered proteins. LLPS is induced by self-association or polymerization of phase-separating biomacromolecules to form droplets, fibers and hydrogels. This mechanism is believed to be a key process in the assembly of membrane-less organelles which control multiple cellular functions. Diverse membrane-less organelles are observed in both the cytoplasm and the nucleus of cells, including stress granules, germ granules, nucleoli, and nuclear speckles. These organelles are composed of multiple proteins and RNA/DNA molecules all demonstrating weak, multivalent interactions with each other [15–19]. A unique component that characterized the membrane-less organelles and is commonly found in them are the RNA binding proteins. The RNA binding protein fused in sarcoma (FUS, also known as translated in liposarcoma/TLS) contains an LC sequence and is prone to self-association. This feature is thought to drive formation of various cellular granules, including stress granules, paraspeckles, and DNA damage foci. Dysfunction of FUS as a result of mutation may cause diverse pathologies including cancer and neurodegenerative diseases like amyotrophic lateral sclerosis (ALS) and fronto-temporal lobar degeneration (FTLD) [20–22]. The localization of FUS to the nucleus is dependent on its import by one of the Kapβ proteins, Kapβ2 (also known as transportin-1). Recent studies have shown that Kapβ2 inhibits FUS mediated LLPS via multiple interactions [23–26]. These findings suggest that Kapβ2 may form part of the regulatory mechanism for the production of membrane-less organelles through its regulation of FUS mediated LLPS.
FUS was originally identified as an oncogene involved in the pathogenesis of liposarcoma [27]. Subsequently, mutations in FUS have been reported in neurodegenerative diseases like ALS and FTLD [22,28]. FUS is encoded by the FUS gene and has been linked to various events within the nucleus including the DNA damage response, transcription, and RNA splicing [29]. It is suggested that abnormal FUS causes several fatal diseases. FUS aggregates are a pathological hallmark of ALS/FTLD-FUS.
FUS is prone to auto-aggregation as a result of its primary structure. FUS is composed of an N-terminal Ser, Tyr, Gly, Gln-rich (SYGQ-rich) region, and three Arg-Gly-Gly (RGG1-3) repeats. There are two folded regions, an RNA recognition motif (RRM), and a Zn-finger (ZnF) motif between the RGG repeats. In addition, FUS possesses a proline-tyrosine nuclear localization signal (PY-NLS) which is recognized by Kapβ2 and which is located at the C-terminus (Fig. 1a) [30]. FUS forms amyloid-like fibril polymers, hydrogels, and droplets via LLPS (Fig. 1b). Biophysical analysis undertaken using various techniques including solution NMR, solid state NMR, hydrogel binding assays, confocal/electron microscopy, and atomic force microscopy have revealed the various physical properties of this protein and its phase-separate/aggregate structures. Because of its high degree of characterization, FUS is commonly used as a reference model for this field of research.
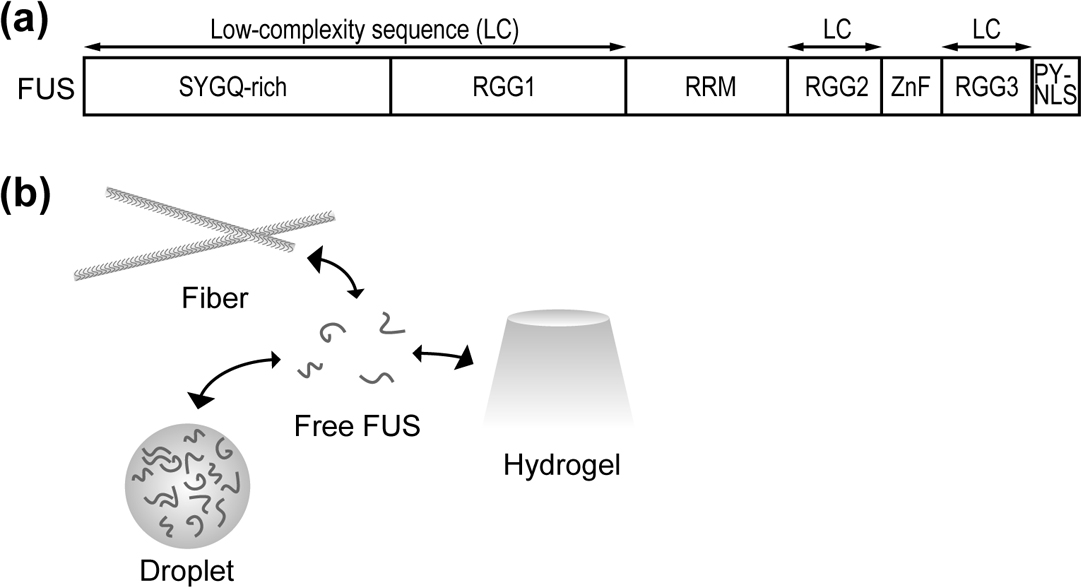
Domain architecture of the FUS protein and its phase separation state. (a) Domain architecture of FUS. (b) Multivalent interactions drive liquid–liquid phase separation. Products of LLPS can be classified as droplets, amyloid-like fibers, and hydrogels. Common features of these products are flexibility and rapid molecular exchange between the interior and exterior of the structure. The mechanism regulating the transition between different states remains undefined.
In 2012, Steven McKnight and his group first reported the hydrogel property of FUS and attributed it to labile cross-β polymers [31]. Subsequent solid-state NMR analysis revealed the cross-β structure of the N-terminal SYGQ-rich LC [32]. Other groups revealed additional physical properties of FUS, including the fact that the RGG repeats do not exhibit phase separation on their own but do so in the presence of the SYGQ-rich LC. Mutational studies revealed that cation-π interactions between tyrosine in the SYGQ-rich LC and arginine in the RGG repeats is important for the phase separation properties of the FUS protein [33]. Post-translational modifications including serine phosphorylation and arginine methylation impair FUS LLPS. Point mutation G156E, one of the ALS-associated mutations in the SYGQ-rich LC domain, accelerates aberrant fibrillation of FUS [34,35]. Protein concentration, salt, nucleic acids, crowding reagents, and alcohol further affect FUS LLPS [11,36].
Signal-mediated nuclear localization is facilitated by nuclear import receptors belonging to the Kapβ protein family. There are 19 known human Kapβ proteins all with similar molecular weight and helical topologies. These topologies are characterized by HEAT repeats, and each Kapβ recognizes a unique nuclear localization signal (NLS) within their cargo in order to facilitate recognition and nuclear import. In the nucleus, GTP-bound small GTPase Ran (RanGTP) dissociates the Kapβ-cargo complex releasing the cargo compounds [3,37,38].
Many of these cargo molecules include a consensus sequence in their NLSs, which identifies them as Importin β (Impβ, also known as Karyopherin β1) and Kapβ2 targets. Impβ binds classical NLS (lysine rich) motifs through an adapter protein, Importin α (Impα, also known as Karyopherin α) and were among the first NLS motifs to be described in the literature [39]. While Kapβ2 binds the PY-NLS directly. PY-NLSs consist of a C-terminal RX2-5PY (X can be any amino acid) motif and an N-terminal short hydrophobic or basic segment motif [40,41]. Many Kapβ2 cargos are RNA binding proteins including FUS and hnRNPs. These proteins play important roles in RNA processing and DNA damage responses in the nuclear puncta. Mis-localization of these RNA binding proteins has been described as a hallmark for several pathologies including cancer and neurodegenerative diseases [42,43]. Recent studies have revealed that these RNA binding proteins form droplets or amyloid-like filaments via LLPS mediated by their LC regions. Therefore, we can assume that cells use LLPS of these RNA binding proteins to create functional compartments and that these LC sequences may control the creation of these structures, and it should be regulated properly.
The cargo of Kapβ2 are highly prone to self-association as a result of their LC sequences. The fundamental question is how Kapβ2 prevents this during nuclear transport? We found that FUS droplets formed via LLPS were dissolved when Kapβ2 was added [23]. This result suggests that Kapβ2 functions as a chaperone to prevent self-association of FUS by suppressing LLPS in the cytoplasm. This chaperone function of Kapβ2 is dependent on the binding of Kapβ2 to FUS through its PY-NLS. FUS-Kapβ2 complexes were crystallized and the structure was elucidated. The regions outside of the PY-NLS of the FUS protein were structurally disordered in the crystal structures indicating that interactions between FUS and Kapβ2 are weak and unstable beyond the PY-NLS (Fig. 2a). NMR detected multiple weak interactions between Kapβ2 and regions of the FUS protein that fall outside of the PY-NLS. Although the PY-NLS of FUS itself does not contribute to LLPS, the tight binding of PY-NLS to Kapβ2 allows weak multiple interactions with protein regions outside of the PY-NLS, including the SYGQ-rich region and RGG repeats (Fig. 2b). This chaperone function is presumably conserved among Kapβs since both Impβ and its yeast homolog karyopherin 121p suppressed FUS LLPS with the help of the PY-NLS sequence. Four different groups reported novel functions for Kapβ proteins in 2018 [23–26].
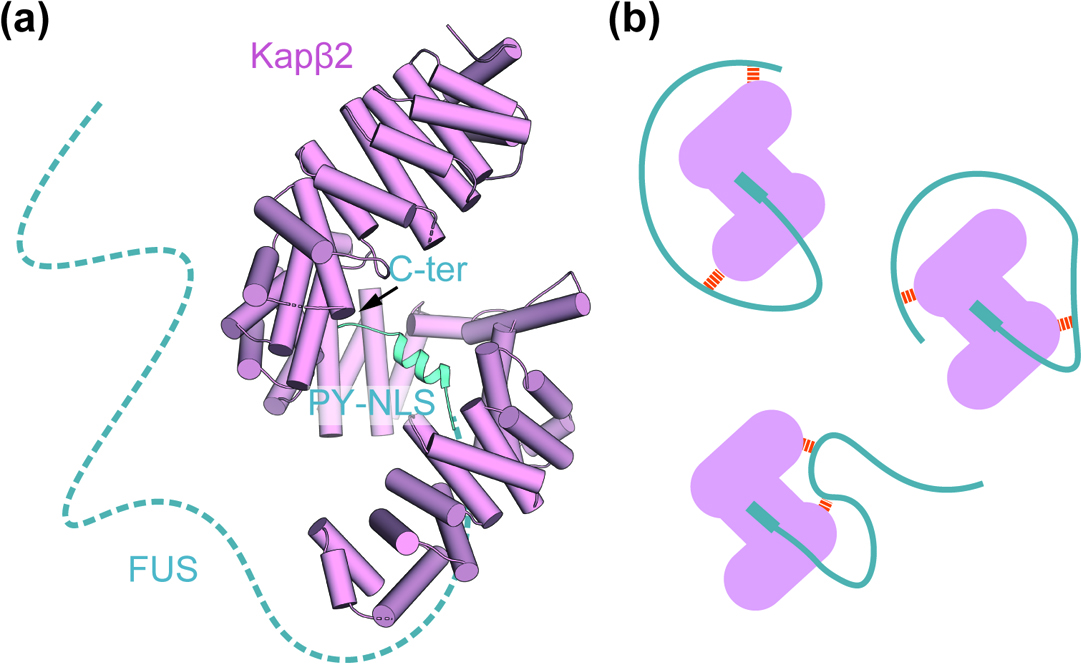
Kapβs block LLPS through multiple interactions. (a) Crystal structure of the FUS-Kapβ2 complex (PDB ID 5YVG). Kapβ2 and FUS are colored in pink and cyan, respectively. FUS model represents only the residues 507–526 in the PY-NLS. Protein regions outside the PY-NLS, which are missing from the crystal structure, are represented by a dashed line. (b) Kapβ2 demonstrates a tight interaction with the PY-NLS region and weak/transient interactions with the regions outside of the FUS PY-NLS, preventing phase separation. Weak/transient interactions are colored in orange.
Kapβ2 interacts with not only the LC regions of the cargo but also the LC regions of the nucleoporins (Nups), components of the nuclear pore complex (NPC) [44–46]. NPC is composed of multiple copies of 30 different Nups. One third of these are composed of an inner ring formed by phenylalanine-glycine repeats (FG-domain). FG-domains are known to form hydrogels via LLPS, which create a permeability barrier to prevent unexpected diffusion of biological macromolecules [47]. Kapβs pass through the barrier by interacting with these FG-repeats. This phenomenon is similar to the dissolution of FUS droplets. Thus, Kapβ proteins use the dissolving function of LLPS during various cellular events.
Several recent publications on LLPS uncovered the importance of droplets/puncta formed via LLPS in eukaryotic cells. Kapβs are not only nuclear transport receptors but also key regulators of the formation of specific droplets. Kapβs may control phase separation states by chaperoning component proteins in order to adapt to environmental changes (Fig. 3). It should be noted that impaired RNA granule formation leads to fatal neurodegenerative diseases. Mutations in FUS family protein hnRNP, from Ewing sarcoma, were found to be associated with familial ALS. Many mutations found in the PY-NLS region, impair interactions with Kapβ2, indicating that aberrations in the precise localization and/or chaperoning activity of Kapβs are important in the pathogenesis of neurodegenerative diseases. Stress granule inducers disrupt nuclear import through incorporation of Kapβs into stress granules. The mechanisms underlying these toxic effects remain unclear. Further studies are needed to establish their molecular basis.
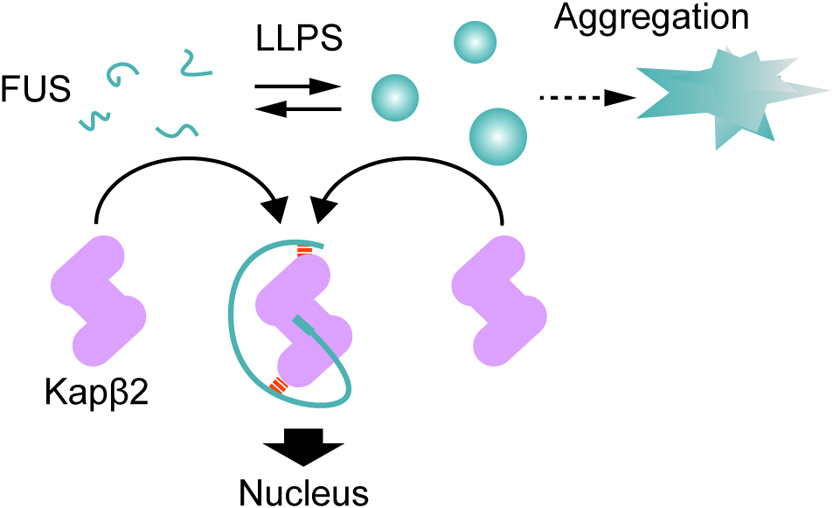
Model of cellular FUS states and their regulation by Kapβ2. Kapβ2 carries newly translated cargo into the nucleus by binding to its specific NLS with high-affinity. During stress, FUS accumulates in the cytoplasm and forms droplets which eventually result in the assembly of the stress granules. Kapβ2 can dissociate these FUS droplets following its interaction with the FUS NLS domain, this suggests that Kapβ proteins may exert a chaperone function preventing phase separation during nuclear transport.
Understanding the roles of molecular chaperones for phase separating proteins is important especially in the generation of novel therapeutics for protein aggregation diseases. Heat shock proteins (HSPs) have also been identified as chaperone candidates for phase separated proteins because many of HSPs are known molecular chaperones of other aggregating proteins. Yeast Hsp104, a hexameric protein disaggregase from the AAA+ ATPase family was reported to disaggregate disordered proteins including FUS [48]. Although Hsp104 does not exist in metazoans, other disaggregase HSPs from humans including HSP110, HSP70, and HSP40 may function in a similar way for other phase separated proteins. Chaperone function of HSPs is ATP dependent and targets a variety of aggregating proteins. By contrast, Kapβ proteins do not require enzymatic activity and target proteins with specialized NLS motifs. Further studies will reveal the chaperones and their functions with respect to various phase separating proteins and provide insights into how LLPS in membrane-less organelles are regulated. Understanding this regulation could present novel avenues for the development of therapeutics used to treat protein aggregation diseases.
This work was supported by the Daiichi Sankyo Foundation for Life Science Grants-in-Aid and The Nakabayashi Trust for ALS Research Grants-in-Aid of T. Y.
The authors have no conflicts of interest to declare.
T. Y. wrote the original draft. H. M. reviewed the manuscript. T. Y. and H. M. edited the manuscript.